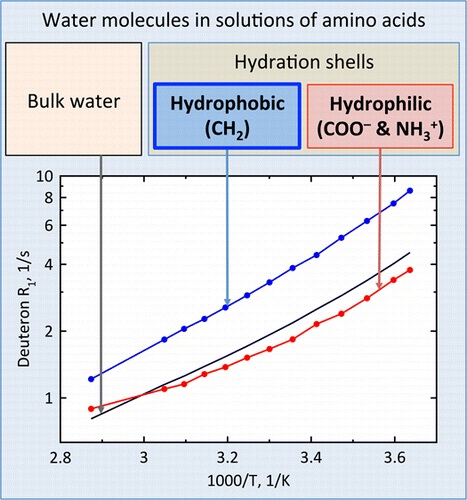
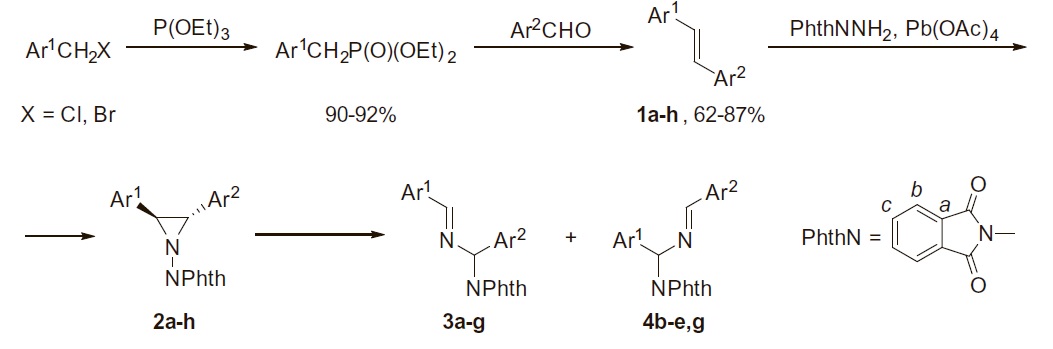
S.N. Britvin, S.A. Kashtanov, M.G. Krzhizhanovskaya, A.A. Gurinov, O.V. Glumov, S. Strekopytov, Yu.L. Kretser, A.N. Zaitsev, N.V. Chukanov, S.V. Krivovichev
“Perovskites with the Framework-Forming Xenon”
Angew. Chem. Int. Ed., 2015, 54, 1-6
DOI:10.1016/j.tet.2015.07.071
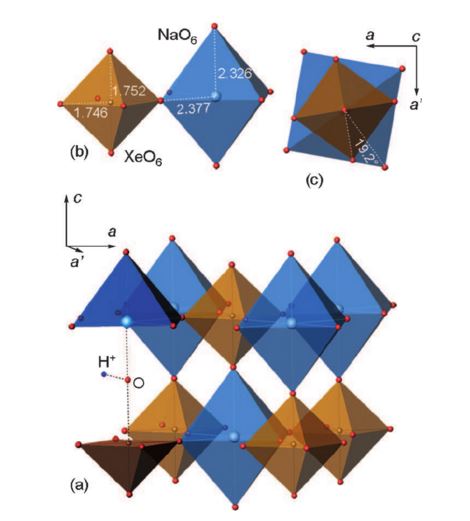
The Group 18 elements (noble gases) were the last ones in the periodic system to have not been encountered in perovskite structures. We herein report the synthesis of a new group of double perovskites KM(XeNaO6) (M=Ca, Sr, Ba) containing framework-forming xenon. The structures of the new compounds, like other double perovskites, are built up of the alternating sequence of corner-sharing (XeO6) and (NaO6) octahedra arranged in a three-dimensional rocksalt order. The fact that xenon can be incorporated into the perovskite structure provides new insights into the problem of Xe depletion in the atmosphere. Since octahedrally coordinated XeVIII and SiIV exhibit close values of ionic radii (0.48 and 0.40 Å, respectively), one could assume that XeVIII can be incorporated into hyperbaric frameworks such as MgSiO3 perovskite. The ability of Xe to form stable inorganic frameworks can further extend the rich and still enigmatic chemistry of this noble gas.