2017 |2016 | 2015 | 2014 | 2013 | 2013-2012
| № | Abstract | |
| 1 | A.M. Afanasenko, E.Yu. Bulatov T.G. Chulkova, Matti Haukka, F. M. Dolgushin, “An efficient method for selective oxidation of (oxime) Pt(II) to (oxime) Pt(IV) species using N,N-dichlorotosylamide”, Transition Met Chem (2016) 41:387–392 DOI:10.1007/s11243-016-0034-7 | |
 |
Abstract.The oxidation of (oxime)PtII species using the electrophilic chlorine-based oxidant N,N-dichlorotosylamide (4-CH3C6H4SO2NCl2) was studied. The reactions of trans-[PtCl2(oxime)2] (where oxime = acetoxime, cyclopentanone oxime, or acetaldoxime) with this oxidant led to trans-[PtCl4(oxime)2] products. The oxidation of trans-[Pt(o-OC6H4CH = NOH)2] at room temperature gave trans-[PtCl2(o-OC6H4CH = NOH)2], whereas the same reaction upon heating was accompanied by electrophilic substitution of the benzene rings. | |
| 2 | Tatyana B. Anisimova, Mikhail A. Kinzhalov, Maxim L. Kuznetsov, M. Fátima C. Guedes da Silva, Andrey A. Zolotarev, Vadim Yu. Kukushkin, Armando J. L. Pombeiro, Konstantin V. Luzyanin, “1,3-Dipolar Cycloaddition of Nitrones to Gold(III)-Bound Isocyanides”, Organometallics 2016, 35, 3569−3576 DOI:10.1021/acs.organomet.6b00635 | |
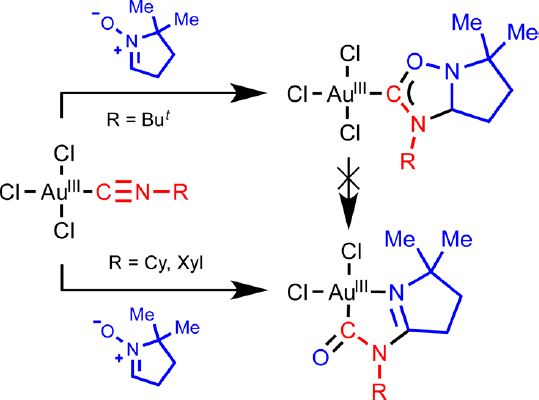 |
Abstract.Treatment of gold(III)-isocyanides [AuCl3(CNR1)] (R1 = Xyl 1, Cy 2, But 3) with an equimolar amount of 5,5-dimethyl-1-pyrroline-N-oxide (4) in CH2Cl2 at −74 °C leads to the generation of the heterocyclic aminocarbene species [AuCl3{C(ONaCMe2CH2CH2CbH)═NeR1}(Na–Cb)(Cb–Ne)] 8 (for R1 = But) or gold(III) complexes cis-[AuCl2{Na(CMe2CH2CH2CbNeR1)Cd═O}(Na═Cb)(Ne–Cd)] 9 and 10 (for R1 = Xyl and Cy) in good isolated yields (75–87%). DFT calculations show that deprotonation of the endocyclic CH group in the carbene ligand leads to spontaneous N–O bond cleavage, and acidity of this group is a factor controlling the different chemical behavior of 1–3 depending on the nature of substituent R1. The reaction of equimolar amounts of the aldonitrone p-TolCH═N+(Me)O– (5) or the ketonitrones Ph2C═N+(R2)O– (R2 = Ph 6, CH2Ph 7) with 1–3 in CD2Cl2 at −70 °C in air (or under N2) revealed the formation of the carbene complexes [AuCl3{C(ONMeCaH-p-Tol)═NbR1}(Ca–Nb)] (R1 = Cy 11, Xyl 12, But 13), [AuCl3{C(ONPhCaPh2)═NbR1}(Ca–Nb)] (R1 = Cy 14, But 15), or [AuCl3{C(ON(CH2Ph)CaPh2)═NbR1}(Ca–Nb)] (R1 = Cy 16, Xyl 17), as studied by 1H NMR. The reaction of 6 with 1 and of 7 with 3 did not furnish carbene products. Compounds 8–10 were characterized by ESI-MS, IR, 1D (1H, 13C{H}) and 2D (1H,1H–COSY, 1H,13C-HSQC, 1H,13C-HMBC) NMR spectroscopic techniques, and, only for 8, elemental analyses (C, H, N), while compounds 11–17 were characterized by 1D (1H, 13C{H}) and 2D (1H,13C-HSQC) NMR. Structures of compounds 8, 9, and 13 were additionally established by single-crystal X-ray diffraction. | |
| 3 | A. Ya. Bespalova, T. L. Gorchakovaa, A. Yu. Ivanovb, M. A. Kuznetsovb, L. M. Kuznetsovab, A. S. Pan’kovab, L. I. Prokopenkoa, and A. F. Khlebnikovb “On the Possibility for Synthesizing Dihydrotriazolothiadiazoles by Condensation of 4-Amino-2,4-dihydro-3H-1,2,4-triazole-3-thiones with Aromatic Aldehydes”, RUSSIAN JOURNAL OF ORGANIC CHEMISTRY Vol. 52 No. 3 2016 DOI:10.1134/S1070428016030210 | |
 |
Abstract.Regardless of the conditions, the condensation of 4-amino-2,4-dihydro-3H-1,2,4-triazole-3-thiones with aromatic aldehydes afforded the corresponding hydrazones as the only product. Both initial amines and resulting hydrazones exist as the thione rather than thiol tautomer. In no case bicyclic 5,6- ihydro[1,2,4]triazolo[3,4-b][1,3,4]thiadiazoles that are isomeric to the hydrazones were detected. DFT quantum chemical calculations at the B3LYP/6-31+g(d,p) level of theory with full geometry optimization showed that the hydrazone structure in methanol and DMF is more stable than the bicyclic isomer by 19–23 kcal/mol, which completely excluded the possibility for such cyclization. The thione tautomer of the hydrazones is more stable than the thiol structure by 11–13 kcal/mol | |
| 4 | Dmitrii S. Bolotin, Alexander S. Novikov, Ilya E. Kolesnikov, Vitaliy V. Suslonov, Yuri Novozhilov, Oksana Ronzhina, Mikhail Dorogov, Mikhail Krasavin, and Vadim Yu. Kukushkin “Phosphorescent Platinum(II) Complexes Featuring Chelated Acetoxime Pyrazoles: Synthetic, Structural, and Photophysical Study”, ChemistrySelect 2016, 3, 456–461 DOI:10.1002/slct.201600130 | |
 |
Abstract.Treatment of cis-[PtCl2(Me2SO)2] with the pyrazole acetoximes p-RC6H4{CN(H)NCCH}CH2C(Me)=NOH (R=OMe, H, F, Cl, CN, NO2),prepared by a cascade reaction from the vinyl isoxazoles p-RC6 H4C(H)=C(H){CONCC(NO2)}Me, leads to the complexes [PtCl2(Ar{CN(H)NCCH}CH2C(Me)=NOH)] (71–88%). The complexes exhibit solid state room temperature phosphorescence with emission maxima in the region of 644–691 nm and biexponential relaxation with t1 ! 2 ms and t2 ! 0.32 ms. In the platinum compounds, substituents R affect phosphorescence quantum yields, which increase by one order of magnitude on going from electron acceptor (R=NO2; 0.2%) to electron-donor groups (R=OMe; 2.0%). Based upon theoretical TD-DFT calculations using the CAM”B3LYP functional, the absorption spectra were attributed to metal-to-ligand charge transfer (1MLCT) and ligand-to-ligand charge transfer (1LLCT) processes. The pyrazole acetoximes and their complexes were characterized by elemental analyses (C, H, N), HRESI-MS, FTIR, 1H NMR (for metalfree species), and CP-MAS TOSS 13C{1H} NMR (for the platinum complexes). In addition, two complexes were studied by singlecrystal X-ray diffraction. |
|
| 5 | D.S. Bolotin, A.S. Novikov, V.K. Burianova, M.Ya. Demakova, E.A. Daines, С. Pretorius, P. Mokolokolo, A. Roodt, N.A. Bokach, M.S. Avdontceva, A.P. Zhdanov, K.Yu. Zhizhin, N.T. Kuznetsov, V.Yu. Kukushkin “Nucleophilicity of oximes based upon addition to а nitrilium closo-decaborate cluster”, Organometallics, 2016, 35, 3612-3623 DOI:10.1021/acs.organomet.6b00678 | |
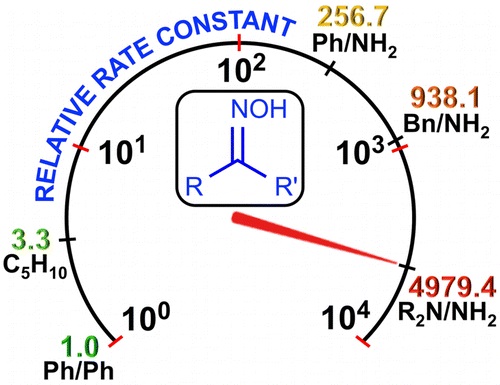 |
Abstract.Three types of oxime species, i.e., 4-morpholylcarbamidoxime (hydroxyguanidine), phenylacetamidoxime and benzamidoxime (amidoximes), and cyclohexanone oxime and benzophenone oxime (ketoximes), react at room temperature with the 2-nitrilium closo-decaborate clusters, leading to 2-iminium closo-decaborates (14 examples; 57–94%). These species were characterized by ICPMS-based boron elemental analysis, HRESI–-MS, molar conductivity, IR, 1H{11B}, and 11B{1H} NMR spectroscopies, and additionally by single-crystal X-ray diffraction (for six compounds). On the basis of kinetic data, ΔH⧧, ΔS⧧, and ΔG⧧ of the additions were determined, showing a 4 order-of-magnitude decrease in reactivity from the hydroxyguanidine to the aromatic ketoxime as entering nucleophiles. The results of DFT calculations indicate that the mechanism for these reactions is stepwise and is realized through the formation of the orientation complex of the nitrone form, R2R3C═N+(H)O–, of oximes with [B10H9NCEt]−, giving further an acyclic intermediate (the rate-determining step), followed by proton migration, leading to the addition product. The calculated overall activation barrier for these transformations is consistent with the experimental kinetic observations. This work provides, for the first time, a broad nucleophilicity series of oximes, which is useful to control various nucleophilic additions of oxime species. | |
| 6 | Natalia A. Danilkina, Larisa Y. Gurskaya, Aleksander V. Vasilyev, and Irina A. Balova “Towards Isocoumarin-Fused Enediyne Systems through the Electrophilic Cyclization of Methyl o-(Buta-1,3-diynyl)benzoates”, Eur. J. Org. Chem. 2016, 739–747 DOI:10.1002/ejoc.201501262 | |
 |
Abstract.The synthesis of enediynes fused to an isocoumarin core was achieved through the electrophilic cyclization of o-(buta-1,3-diynyl)benzoates as a key step, followed by the Sonogashira coupling of the resulting 3-ethynyl-4-iodoisocoumarins with acetylenes. This approach allowed different substituents to be introduced to both ethynyl moieties with complete regiocontrol. The ability of the isocoumarin-fused enediynes to undergo Bergman cyclization was studied by differential scanning calorimetry. | |
| 7 | E. Ejarque and E. Abakumov “Stability and biodegradability of organic matter from Arctic soils ofWestern Siberia: insights from 13C-NMR spectroscopy and elemental analysis”, Solid Earth, 7, 153–165, 2016 DOI: 10.5194/se-7-153-2016 | |
 |
Abstract.Arctic soils contain large amounts of organic matter which, globally, exceed the amount of carbon stored in vegetation biomass and in the atmosphere. Recent studies emphasise the potential sensitivity for this soil organic matter (SOM) to be mineralised when faced with increasing ambient temperatures. In order to better refine the predictions about the response of SOM to climate warming, there is a need to increase the spatial coverage of empirical data on SOM quantity and quality in the Arctic area. This study provides, for the first time, a characterisation of SOM from the Gydan Peninsula in the Yamal Region,Western Siberia, Russia. On the one hand, soil humic acids and their humification state were characterised by measuring the elemental composition and diversity of functional groups using solidstate 13C-nuclear magnetic resonance (NMR) spectroscopy. Also, the total mineralisable carbon was measured. Our results indicate that there is a predominance of aliphatic carbon structures, with a minimal variation of their functional-group composition both regionally and within soil depth. This vertical homogeneity and low level of aromaticity reflects the accumulation in soil of lowly decomposed organic matter due to cold temperatures. Mineralisation rates were found to be independent of SOM quality, and to be mainly explained solely by the total carbon content. Overall, our results provide further evidence on the sensitivity that the soils ofWestern Siberia may have to increasing ambient temperatures and highlight the important role that this region can play in the global carbon balance under the effects of climate warming. |
|
| 8 | Edward I. Evstigneyev, Oleg S. Yuzikhin, Andrey A. Gurinov, Alexandr Yu. Ivanov, Tatiana O. Artamonova, Mikhail A. Khodorkovskiy, Elena A. Bessonova & Aleksander V. Vasilyev “Study of Structure of Industrial Acid Hydrolysis Lignin, Oxidized in the H2O2-H2SO4 System”, Journal of Wood Chemistry and Technology, 36 (2016), 259–269, DOI: 10.1080/02773813.2015.1137945 | |
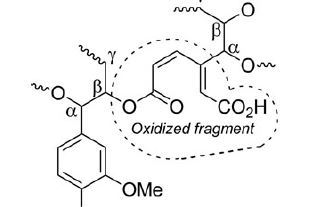 |
Abstract.Products of oxidation of industrial acid hydrolysis lignin in the H2O2-H2SO4 system were studied using 13C NMR (in solution and solid state), MALDI-MS, and MS(ESI) techniques. Oxidation of hydrolysis lignin leads to the opening of aromatic rings of lignin, yielding carboxylic groups. Alkyl aryl ether linkages (β-O-4-bonds) between lignin phenyl propane units are not significantly affected by the oxidation. The structure of oxidized hydrolysis lignin is proposed. The basic structural unit of oxidized hydrolysis lignin is a muconic acid derivative. | |
| 9 | Liya D. Funt, Olesya A. Tomashenko, Alexander F. Khlebnikov, Mikhail S. Novikov, and Alexander Yu. Ivanov “Synthesis, Transformations of Pyrrole- and 1,2,4-Triazole-Containing Ensembles, and Generation of Pyrrole-Substituted Triazole NHC”, J. Org. Chem. 2016, 81, 11210−11221, DOI: 10.1021/acs.joc.6b02200 | |
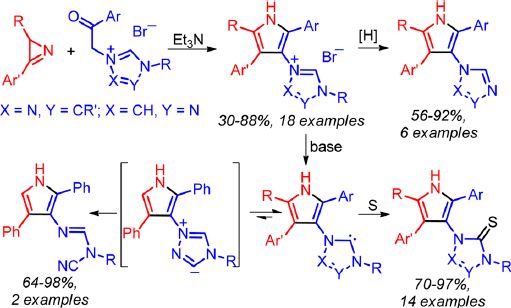 |
Abstract.Unprecedented pyrrole- and 1,2,4-triazole-containing ensembles, substituted 1-(1H-pyrrol-3-yl)-4H-1,2,4-triazol-1- ium bromides and 4-(1H-pyrrol-3-yl)-1H-1,2,4-triazol-4-ium bromides, were prepared from 2H-azirines and triazolium phenacyl bromides using a simple procedure. N-(1H-Pyrrol-3-yl)-N′-benzyltriazolium bromides undergo reductive debenzylation on Pd/C to give substituted 1-(1H-pyrrol-3-yl)-4H-1,2,4-triazoles and 4-(1H-pyrrol-3-yl)-1H-1,2,4-triazoles in high yields. Betaines (triazoliumylpyrrolides) and pyrrolyltriazole NHCs, which are possible products of dehydrobromination of pyrrolyltriazolium salts, have comparable thermodynamic stabilities in nonpolar solvents according to calculations at the DFT B3LYP/6-31G(d) level. The carbene forms can be easily trapped by the reaction of salts with base in the presence of sulfur. The corresponding 1- and 4-(1H-pyrrol-3-yl)-1H-1,2,4-triazole-5(4H)-thiones are formed in high yields. In the absence of sulfur as a trap, the opening of the triazole ring occurs with the formation of derivatives of N-cyanoformimidamide. According to the DFT calculations the latter is most probably formed via a pyrrolyltriazoliumide intermediate, which is the minor component of the equilibrium triazoliumylpyrrolide−pyrrolyltriazole NHC−pyrrolyltriazoliumide. Blocking of the pyrrolyltriazoliumide intermediate formation, by introduction of a substituent at the 3-position of the triazole ring, made it possible to generate the first pyrrole-substituted triazole NHC. | |
| 10 | Daniil M. Ivanov, Alexander S. Novikov, Ivan V. Ananyev, Yulia V. Kirinaa and Vadim Yu. Kukushkin “Halogen bonding between metal centers and halocarbons”, Chem. Commun., 2016, 52, 5565-5568, DOI: 10.1039/c6cc01107a | |
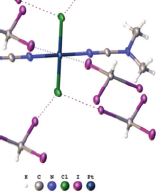 |
Abstract.Metal-involving halogen bonding was detected in a series of associates of CHI3 with trans-[PtX2(NCNAlk2)2] (X = Cl, Br). The HI2C–I…n1 (Pt) halogen bonding and the bifurcated HI2C–I…n2 (Pt–Cl) halogen bonding – the latter undergoes the thermally induced reversible HI2C–I…n2 (Pt–Cl) <=> HI2C–I…n1 (Pt) transformation – were observed and confirmed theoretically. | |
| 11 | Daniil M. Ivanov, Pavel V. Gushchin, Alexander S. Novikov, Margarita S. Avdontceva, Andrey A. Zolotarev, Galina L. Starova, Yi-Ting Chen, Shih-Hung Liu, Pi-Tai Chou, and Vadim Yu. Kukushkin “Platinum(II)-Mediated Double Coupling of 2,3-Diphenylmaleimidine with Nitrile Functionalities To Give Annulated Pentaazanonatetraenate (PANT) Systems”, Eur. J. Inorg. Chem. 2016, 1480–1487, DOI: 10.1002/ejic.201501398 | |
 |
Abstract.Treatment of trans-[PtCl2(NCR)2] [R = Et 1, nPr 2, tBu 3, CH2Ph 4, Ph 5, p-CF3C6H4 6, NMe2 7, NEt2 8, N(CH2)5 9] with 2.5 equiv. of 2,3-diphenylmaleimidine in CH2Cl2 at room temperature for 5 min [for R = p-CF3C6H4 6, NMe2 7, NEt2 8, N(CH2)5 9] or 14 h (for R = Et 1, nPr 2, tBu 3, CH2Ph 4, Ph 5) furnishes (1,3,5,7,9-pentaazanona-1,3,6,8-tetraenato)platinum(II) [(PANT)PtII; PtCl{HN=C(R)N=CN[C(Ph)=C(Ph)]C=NC(R)=NH}] complexes 10–18. These species are formed by platinum(II)-mediated double coupling of 2,3-diphenylmaleimidine with both nitrile ligands. The formulation of the complexes was supported by satisfactory C, H, and N elemental analyses, which were in agreement with HRESI-MS, IR, and 1H and 13C{1H} NMR spectra. The structures of 10, 11·1/8nC6H14, 15, 16·CCl4, and 17·CHCl3 were determined by single-crystal X-ray diffraction. Absorption and emission studies were performed on representative complexes 10, 15, and 18, and the results show weak phosphorescence maxima at around 730–750 and 770–820 nm in CH2Cl2 and in the solid state, respectively. Further insight into the photophysical properties was gained by time-dependent density functional theory (TD–DFT) with detailed analysis of the corresponding frontier molecular orbitals for the lower-lying transition. The calculated energies of the T1 state (in terms of wavelength) are 715.8 nm for 10, 764.6 nm for 15, and 697.7 nm for 18, and these values are in good agreement with the trend of the first vibronic peaks of their phosphorescence spectra. |
|
| 12 | Daniil M. Ivanov, Yulia V. Kirina, Alexander S. Novikov, Galina L. Starova, Vadim Yu. Kukushkin “Efficient p-stacking with benzene provides 2D assembly of trans-[PtCl2(p-CF3C6H4CN)2]”, Journal of Molecular Structure 1104 (2016) 19e23, DOI: 10.1016/j.molstruc.2015.09.027 | |
 |
Abstract.The new platinum(II) complex trans-[PtCl2(p-CF3C6H4CN)2], which was obtained upon treatment of [PtCl2(MeCN)2] with excess of p-CF3C6H4CN, co-crystallizes with benzene giving 2D layers formed by complexebenzene and complexecomplex p-stacking interactions. These interactions were detected by X-ray crystallography and their nature was analyzed by DFT calculations (M06 functional), including AIM analysis, inspection of NBO atomic charges, and evaluation of the vertical total energies for dissociation in the model supramolecular cluster. The crystal packing insignificantly effect the structure of the supramolecular associates and p-stacking interactions are rather weak. | |
| 13 | Е. А. Катленок, А. А. Золотарев, А. Ю. Иванов, С. Н. Смирнов, Р. И. Байчурин, К. П. Балашев “СТРОЕНИЕ, ОПТИЧЕСКИЕ И ЭЛЕКТРОХИМИЧЕСКИЕ СВОЙСТВА КОМПЛЕКСОВ Ir(III) И Pt(II) С ЦИКЛОМЕТАЛЛИРОВАННЫМ 2-ФЕНИЛБЕНЗОТИАЗОЛОМ И ХЕЛАТИРУЮЩИМИ ДИЭТИЛДИТИОКАРБАМАТ- И О-ЭТИЛДИТИОКАРБОНАТ-ИОНАМИ”, КООРДИНАЦИОННАЯ ХИМИЯ, 2016, том 42, № 3, с. 156–163, DOI: 10.7868/S0132344X16030038 | |
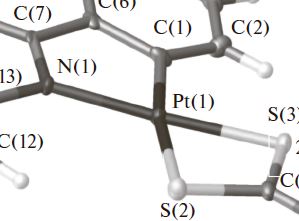 |
Abstract.Методами РСА, ИК и ЯМР 1Н, 13С{1H}, 195Pt спектроскопии показано квадратное строение комплексов Pt(II) и цис-С,С-строение октаэдрических комплексов Ir(III) с металлированным 2-фенилбензотиазолом и хелатирующими диэтилдитиокарбамат- и О-этилдитиокарбонат-ионами. Высшая заполненная и низшая свободная молекулярные орбитали комплексов, определяющие их длинноволновое поглощение и фосфоресценцию, одноэлектронное окисление и восстановление, отнесены к преимущественно локализованным на смешанных p(S)/d(M) и π*-орбиталях металлированного лиганда. Катодное смещение вольтамперограммы окисления и батохромное смещение фосфоресценции комплекса Pt(II) c О-этилдитиокарбонат-ионом отнесены к повышенному донорно-акцепторному взаимодействию донорных атомов S лиганда c Pt(II). | |
| 14 | E. A. Katlenok, A. A. Zolotarev, A. Yu. Ivanov, S. N. Smirnov, R. I. Baichurin, and K. P. Balashev “Complexes of Ir(III) and Pt(II) with Cyclometallated 2-Phenylbenzothiazole and Chelating Diethyldithiocarbamate and O-Ethyldithiocarbonate Ions: Structures and Optical and Electrochemical Properties”, RUSSIAN JOURNAL OF COORDINATION CHEMISTRY 42 (No. 3, 2016) 178–186 DOI: 10.1134/S1070328416030039 | |
|
Abstract.It is shown by X-ray diffraction analysis, IR spectroscopy, and 1Н, 13С{1H}, and 195Pt NMR spectroscopy that the Pt(II) and octahedral Ir(III) complexes with metallated 2-phenylbenzothiazole and chelating diethyldithiocarbamate and O-ethyldithiocarbonate ions have the square and cis-C,C structure, respectively. The highest occupied and lowest unoccupied molecular orbitals of the complexes determining their long-wavelength absorption, phosphorescence, and one-electron oxidation and reduction are assigned to those predominantly localized on the mixed p(S)/d(M) and π* orbitals of the metallated ligand. The cathodic shift of the oxidation voltammogram and the bathochromic phosphorescence shift of the Pt(II) complex with the О-ethyldithiocarbonate ion are attributed to the enhanced donor–acceptor interaction of the donor S atoms of the ligand with Pt(II). The structural data are deposited with the Cambridge Crystallographic Data Centre (CIF files CCDC nos. 1058768 (Ia) and 1058767 (IIb)). |
|
| 15 | Mikhail A. Kinzhalov, Alexander S. Novikov, Konstantin V. Luzyanin, Matti Haukka, Armando J. L. Pombeirod and Vadim Yu. Kukushkin “Complexes of Ir(III) and Pt(II) with Cyclometallated 2-Phenylbenzothiazole and Chelating Diethyldithiocarbamate and O-Ethyldithiocarbonate Ions: Structures and Optical and Electrochemical Properties”, New J. Chem., 2016, 40, 521-527 DOI: 10.1039/c5nj02564h | |
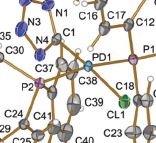 |
Abstract.Reaction between equimolar amounts of trans-[PdCl(PPh3)2(CNR)][BF4] (R = t-Bu 1, Xyl 2) and diisopropylammonium azide 3 gives the tetrazolate trans-[PdCl(PPh3)2(CN4t-Bu)] (67%, 4) or trans-[PdCl(PPh3)2(CN4Xyl)] (72%, 5) complexes. 4 and 5 were characterized by elemental analyses (C, H, N), HRESI+-MS, 1H and 13C{1H} NMR spectroscopies. In addition, the structure of 4 was elucidated by a single-crystal X-ray diffraction. DFT calculations showed that the mechanism for the formal cycloaddition (CA) of N3 to trans-[PdCl(PH3)2(CNMe)]+ is stepwise. The process is both kinetically and thermodynamically favorable and occurs via the formation of an acyclic NNNCN-intermediate. The second step of the formal CA, i.e. cyclization, is rate limiting. Despite the fact that the substitution of CNMe by the N3 ligand is slightly thermodynamically favorable, we were unable to find paths on the potential energy surface for hypothetical CA between uncomplexed isocyanide and palladium-bound azide. Thus, we believe that the experimentally observed palladium tetrazolate complexes are, in fact, generated from the negatively charged uncomplexed azide and the positively charged metal-bound isocyanide species, and this reaction path is favorable from the viewpoint of Coulomb attraction. |
|
| 16 | Alexander S. Konev, Alexander F. Khlebnikov, Oleg V. Levin, Daniil A. Lukyanov, and Ivan M. Zorin “Photocurrent in Multilayered Assemblies of PorphyrinFullerene Covalent Dyads: Evidence for Channels for Charge Transport”, ChemSusChem 2016, 9, 676 – 686 DOI: 10.1002/cssc.201501686 | |
 |
Abstract. Specially designed porphyrin-fullerene dyads have been synthesized to verify literature predictions based on quantum chemistry calculations that certain porphyrin-fullerene dyads are able to self-arrange into specific structures providing channels for charge transport in a bulk mass of organic compound. According to AFM and SEM data, the newly synthesized compounds were indeed prone to some kind of self-arrangement, although to a lesser degree than was expected. A dispersion |
|
| 17 | Dzhamilya N. Konshina, Victor V. Open’ko, Zaual A. Temerdashev, Andrey A. Gurinov & Valeriy V. Konshin “Synthesis of novel silica-gel-supported thiosemicarbazide and its properties for solid phase extraction of mercury”, SEPARATION SCIENCE AND TECHNOLOGY 51, (NO. 7, 2016) 1103–1111 DOI: 10.1080/01496395.2016.1143005 | |
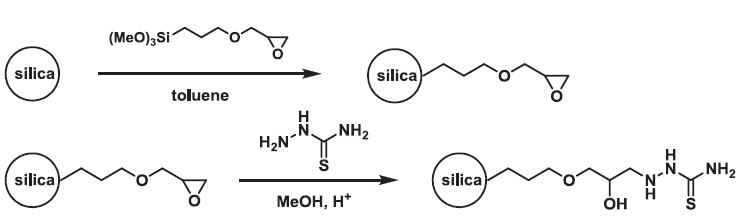 |
Abstract. A new thiosemicarbazidе-modified silica gel (SG-THSC) sorbent was prepared. The sorbent was quantified by adsorption of mercury ions on silica gel, desorption and then spectrophotometry detection of mercury ions. The retention parameters (sample flow rate, eluent type, sample volume, presence of foreign ions, shaking time, sample flow rate and volume, eluent condition, interfering substances) were investigated. The quantitative recovery (>95%) of Hg(II) ions could be obtained by use of 5 mL of 6 mol L−1 HCl. The adsorption capacity of SG-THSC was found to be 98.3 mg g–1 at optimum pH. The maximum preconcentration factor was 400. The technique detection limit was 70 ng L–1, and the relative standard deviation was lower than 4.0% (n = 6). The studied sorbent was applied to preconcentrate the trace Hg(II) fromthe mineralised residues of fish and seawater samples. | |
| 18 | Дж. Н. Коншина, А. В. Данилова, З. А. Темердашев, С. Н. Болотин, А. А. Гуринов, В. В. Коншин “ПОЛУЧЕНИЕ И НЕКОТОРЫЕ СВОЙСТВА СИЛИКАГЕЛЯ С ИММОБИЛИЗОВАННОЙ ФОРМАЗАНОВОЙ ГРУППОЙ”, Журнал прикладной химии Т. 89. (Вып. 4, 2016) | |
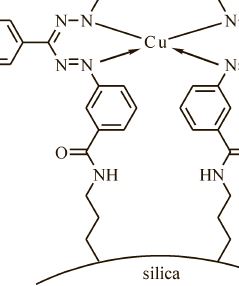 |
Abstract. Предложен и охарактеризован новый сорбционный материал — силикагель с ковалентно иммобилизованной формазановой группой, получаемый сочетанием иммобилизованной соли арилдиазония с фенилгидразоном бензальдегида. Определены некоторые равновесные и кинетические характеристики сорбции Cu(II), Co(II), Ni(II) и Cd(II) из растворов полученным модифицированным силикагелем, показана его эффективность для концентрирования Cu(II) из растворов сложного состава. По данным ЭПР определено координационное окружение Cu(II) в комплексе на поверхности сорбента. | |
| 19 | Kirill I. Kulish, Alexander S. Novikov, Peter M. Tolstoy, Dmitrii S. Bolotin, Nadezhda A. Bokach, Andrey A. Zolotarev, Vadim Yu. Kukushkin “Solid state and dynamic solution structures of O-carbamidine amidoximes gives further insight into the mechanism of zinc(II)-mediated generation of 1,2,4-oxadiazoles”, Journal of Molecular Structure 1111 (2016) 142-150 DOI: 10.1016/j.molstruc.2016.01.038 | |
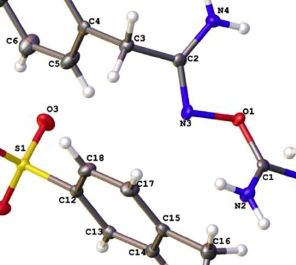 |
Abstract. Three new iminium salts [H2N]C(R)ON]C(R0)NH2](p-TolSO3)$½H2O ([1e3](p-TolSO3)$½H2O; R/R0 ¼ NMe2/PhCH2 1, NMe2/p-BrC6H4 2, N(CH2)5/p-BrC6H4 3) were synthesized via ZnII-mediatedamidoxime-cyanamide coupling and their solid structures were studied by X-ray diffraction. Solution structure and conformational changes of [1e3](p-TolSO3)$½H2O were studied by dynamic NMR. The obtained quantitative data were supported by DFT calculations. All the obtained results help to understand the relative stability of the salts [H2N]C(R)ON]C(R0)NH2](X) (R ¼ NAlk2, Alk, Ar) and give a further insight into the mechanism of ZnII-mediated generation of 1,2,4-oxadiazoles. The electron delocalization and sesquialteral bonds in the [H2N]C(NR2)ON]C(R0)NH2]þ system was recognized by estimation of values of activation energy barriers (14e18 kcal/mol by DNMR and 16e17 kcal/mol by DFT calculations) for the rotation around the CN bonds for the NR2 groups and inspection of the solid-state Xray data along with the Wiberg bond indices (intermediate single/double bond order for the CN distances). This electron delocalization is responsible for the stabilization of the positively charged iminium cation. The moderate strength hydrogen bonding between the oxime N atom and the ¼NH2 group, which is verified from the X-ray, DNMR experiments, and by using quantum chemical calculations, stabilizes the iminium salt, but it is still weak to prevent the heterocyclization. Theoretical calculations of the heterocyclization of [H2N]C(R)ON]C(R0)NH2]þ to 1,2,4-oxadiazoles demonstrated that it is kinetically hindered to a greater extent for R ¼ NAlk2 and this explains their lower reactivity as compared to the iminium salts with R ¼ Alk, Ar. |
|
| 20 | Denis A. Markelov, Vladimir V. Matveev, Petri Ingman, Marianna N. Nikolaeva, Anastasia V. Penkova, Erkki Lahderanta, Natalia I. Boiko & Vladimir I. Chizhik “Unexpected Temperature Behavior of Polyethylene Glycol Spacers in Copolymer Dendrimers in Chloroform”, Scientific Reports 6:24270 DOI: 10.1038/srep24270 | |
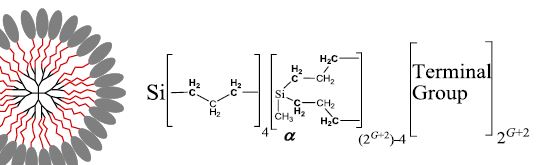 |
Abstract. We have studied copolymer dendrimer structure: carbosilane dendrimers with terminal phenylbenzoate mesogenic groups attached by poly(ethylene) glycol (PEG) spacers. In this system PEG spacers are additional tuning to usual copolymer structure: dendrimer with terminal mesogenic groups. The dendrimer macromolecules were investigated in a dilute chloroform solution by 1H NMR methods (spectra and relaxations). It was found that the PEG layer in G = 5 generations dendrimer is “frozen” at high temperatures (above 260 K), but it unexpectedly becomes “unfrozen” at temperatures below 250 K (i.e., melting when cooling). The transition between these two states occurs within a small temperature range (~10 K). Such a behavior is not observed for smaller dendrimer generations (G = 1 and 3). This effect is likely related to the low critical solution temperature (LCST) of PEG and is caused by dendrimer conformations, in which the PEG group concentration in the layer increases with growing G. We suppose that the unusual behavior of PEG fragments in dendrimers will be interesting for practical applications such as nanocontainers or nanoreactors. | |
| 21 | Jury J. Medvedev, Dmitrii V. Semenok, Xenia V. Azarova, Liudmila L. Rodina, Valerij A. Nikolaev “A New Powerful Approach to Multi-Substituted 3(2H)-Furanones via Brønsted Acid-Catalyzed Reactions of 4-Diazodihydrofuran-3-ones”, Synthesis 2016, 48, A–H DOI: 10.1055/s-0036-1588304 | |
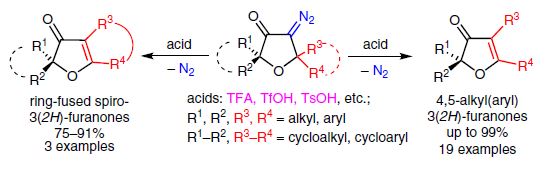 |
Abstract. The interaction of 5,5-dialkyl(diaryl)-substituted 4-diazo-3(2H)furanones with Brønsted acids (TFA, TsOH, etc.) causes elimination of nitrogen accompanied by 1,2-nucleophilic rearrangement, giving rise to exclusive formation of 4,5-dialkyl(diaryl)-substituted 3(2Н)-furanones, ring-fused 3(2H)-furanones, and phenanthro[9,10-b]furan-3(2H)-ones in yields of up to 99%. The reaction is a new highly efficient way for the synthesis of multisubstituted 3(2Н)furanones. | |
| 22 | Alexander S. Mikherdov, Mikhail A. Kinzhalov, Alexander S. Novikov, Vadim P. Boyarskiy, Irina A. Boyarskaya, Dmitry V. Dar’in, Galina L. Starova, and Vadim Yu. Kukushkin “Difference in Energy between Two Distinct Types of Chalcogen Bonds Drives Regioisomerization of Binuclear (Diaminocarbene)PdII Complexes”, J. Am. Chem. Soc. 2016, 138, 14129−14137 DOI: 10.1021/jacs.6b09133 | |
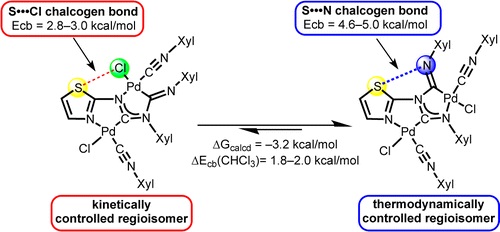 |
Abstract. The reaction of cis-[PdCl2(CNXyl)2] (Xyl = 2,6-Me2C6H3) with various 1,3-thiazol- and 1,3,4-thiadiazol-2-amines in chloroform gives a mixture of two regioisomeric binuclear diaminocarbene complexes. For 1,3-thiazol-2-amines the isomeric ratio depends on the reaction conditions and kinetically (KRs) or thermodynamically (TRs) controlled regioisomers were obtained at room temperature and on heating, respectively. In CHCl3 solutions, the isomers are subject to reversible isomerization accompanied by the cleavage of Pd–N and C–N bonds in the carbene fragment XylNCN(R)Xyl. Results of DFT calculations followed by the topological analysis of the electron density distribution within the formalism of Bader’s theory (AIM method) reveal that in CHCl3 solution the relative stability of the regioisomers (ΔGexp = 1.2 kcal/mol; ΔGcalcd = 3.2 kcal/mol) is determined by the energy difference between two types of the intramolecular chalcogen bonds, viz. S···Cl in KRs (2.8–3.0 kcal/mol) and S···N in TRs (4.6–5.3 kcal/mol). In the case of the 1,3,4-thiadiazol-2-amines, the regioisomers are formed in approximately equal amounts and, accordingly, the energy difference between these species is only 0.1 kcal/mol in terms of ΔGexp (ΔGcalcd = 2.1 kcal/mol). The regioisomers were characterized by elemental analyses (C, H, N), HRESI+-MS and FTIR, 1D (1H, 13C{1H}) and 2D (1H,1H-COSY, 1H,1H-NOESY, 1H,13C-HSQC, 1H,13C-HMBC) NMR spectroscopies, and structures of six complexes (three KRs and three TRs) were elucidated by single-crystal X-ray diffraction. | |
| 23 | Sergey Miltsov, Vladimir Karavan, Alexandr Misharev, Julian Alonso-Chamarro, Mar Puyol “Boron trifluoride–methanol complex. Mild and powerful reagent for deprotection of acetylated amines. Scope and selectivity”, Tetrahedron Letters 57 (2016) 641–644 DOI: 10.1016/j.tetlet.2015.12.087 | |
 |
Abstract. A boron trifluoride–methanol complex demonstrated remarkable deprotection selectivity against commonly used amino-protecting groups in the deacetylation of acetanilides and high sensitivity to the steric hindrance of substrates. The scope and limitations of the reaction were explored. | |
| 24 | Maria S. Mishina, Alexander Yu. Ivanov, Pavel S. Lobanov, Dmitrii V. Dar’in “A New Synthesis of 2-Aminoindoles and 6-Aminopyrrolo[3,2-d]pyrimidines from π-Deficient 1,2-Dihaloarenes and Geminal Enediamines”, Synthesis 48 (2016), 2851–2862 DOI: 10.1055/s-0035-1561645 | |
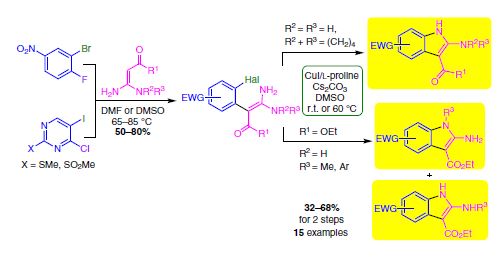 |
Abstract. An efficient approach for the synthesis of fused 2-aminopyrroles via geminal enediamines and π-deficient 1,2-dihaloarenes is presented. The two-step methodology includes aromatic nucleophilic substitution of the activated halogen of dihaloarene with enediamine Cnucleophilic center followed by Cu-catalyzed intramolecular N-arylation. This approach allows access to a variety of 2-amino-6-nitroindoles and 6-aminopyrrolo[3,2-d]pyrimidines (including N-mono- and N,Ndisubstituted) in moderate and good yields under mild conditions. | |
| 25 | A. A. Osipov, V. E. Eremyashev, A. S. Mazur, P. M. Tolstoi, and L. M. Osipov “Coordination State of Aluminum and Boron in Barium Aluminoborate Glass”, Glass Physics and Chemistry, 42 (2016, No. 3) 230–237 DOI: 10.1134/S1087659616030111 | |
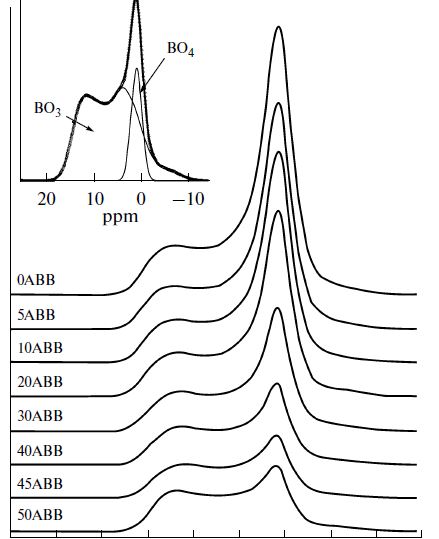 |
Abstract. This paper considers the coordination state of boron and aluminum ions in barium aluminoborate glass with a constant ratio of BaO : B2O3 = 0.5 and a variable ratio of Al2O3 : BaO = 0–3. The dependence of the concentrations of boron and aluminum atoms with a variable coordination number on the Al2O3 content was estimated by IR, 11B and 27Al NMR spectroscopy. The nonlinear nature of the obtained dependences was attributed to variations in the aluminum oxide properties. At a content of less than 30 mol % Al2O3 serves pri marily as a network former, while an increase in the Al2O3 concentration results in its higher modifying role in the studied glass. |
|
| 26 | Yulia S. Panova, Alexey S. Kashin, Maxim G. Vorobev, Evgeniya S. Degtyareva, and Valentine P. Ananikov “Nature of the Copper-Oxide-Mediated C−S Cross-Coupling Reaction: Leaching of Catalytically Active Species from the Metal Oxide Surface”, ACS Catal. 2016, 6, 3637−3643 DOI: 10.1021/acscatal.6b00337 | |
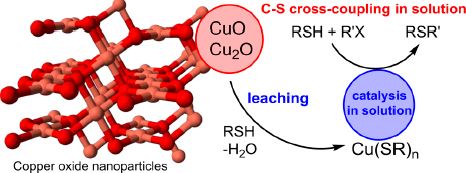 |
Abstract. Copper-oxide-catalyzed cross-coupling reaction is a well-known strategy in heterogeneous catalysis. A large number of applications have been developed, and catalytic cycles have been proposed based on the involvement of the copper oxide surface. In the present work, we have demonstrated that copper(I) and copper(II) oxides served as precursors in the coupling reaction between thiols and aryl halides, while catalytically active species were formed upon unusual leaching from the oxide surface. A powerful cryo-SEM technique has been utilized to characterize the solution-state catalytic system by electron microscopy. A series of different experimental methods were used to reveal the key role of copper thiolate intermediates in the studied catalytic reaction. The present study shows an example of leaching from a metal oxide surface, where the leaching process involved the formation of a metal thiolate and the release of water. A new synthetic approach was developed, and many functionalized sulfides were synthesized with yields of up to 96%, using the copper thiolate catalyst. The study suggests that metal oxides may not act as an innocent material under reaction conditions; rather, they may represent a source of reactive species for solution-state homogeneous catalysis. | |
| 27 | Alexander A. Penney, Vladimir V. Sizov, Elena V. Grachova, Dmitry V. Krupenya, Vladislav V. Gurzhiy, Galina L. Starova, and Sergey P. Tunik “Aurophilicity in Action: Fine-Tuning the Gold(I)−Gold(I) Distance in the Excited State To Modulate the Emission in a Series of Dinuclear Homoleptic Gold(I)−NHC Complexes”, Inorg. Chem. 2016, 55, 4720−4732 DOI: 10.1021/acs.inorgchem.5b02722 | |
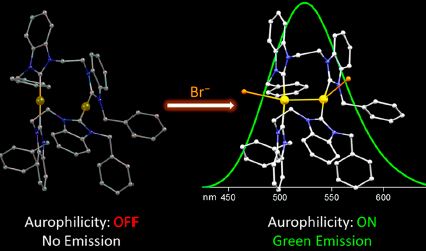 |
Abstract. The solution-state emission profiles of a series of dinuclear Au(I) complexes 4−6 of the general formula Au2(NHC-(CH2)n-NHC)2Br2, where NHC = N-enzylbenzimidazol-2-ylidene and n = 1−3, were found to be markedly different from each other and dependent on the presence of excess bromide. The addition of excess bromide to the solutions of 4 and 6 leads to red shifts of ca. 60 nm, and in the case of 5, which is nonemissive when neat, green luminescence emerges. A detailed computational study undertaken to rationalize the observed behavior revealed the determining role aurophilicity plays in the photophysics of these compounds, and the formation of exciplexes between the complex cations and solvent molecules or counterions was demonstrated to significantly decrease the Au−Au distance in the triplet excited state. A direct dependence of the emission wavelength on the strength of the intracationic aurophilic contact allows for a controlled manipulation of the emission energy by varying the linker length of a diNHC ligand and by judicial choice of counterions or solvent. Such unique stimuli-responsive solution-state behavior is of interest to prospective applications in medical diagnostics, bioimaging, and sensing. In the solid, the investigated complexes are intensely phosphorescent and, notably, 5 and 6 exhibit reversible luminescent mechanochromism arising from amorphization accompanied by the loss of co-crystallized methanol molecules. The mechano-responsive properties are also likely to be related to changes in bromide coordination and the ensuing alterations of intramolecular aurophilic interactions. Somewhat surprisingly, the photophysics of NHC ligand precursors 2 and 3 is related to the formation of groundstate associates with bromide counterions through hydrogen bonding, whereas 1 does not appear to bind its counterions. |
|
| 28 | Elena A. Popova, Tatiyana V. Serebryanskaya, Stanislav I. Selivanov, Matti Haukka, Taras L. Panikorovsky, Vladislav V. Gurzhiy, Ingo Ott, Rostislav E. Trifonov, and Vadim Yu. Kukushkin “Water-Soluble Platinum(II) Complexes Featuring 2-Alkyl-2Htetrazol-5-ylacetic Acids: Synthesis, Characterization, and Antiproliferative Activity”, Eur. J. Inorg. Chem. 2016, 4659–4667 DOI: 10.1002/ejic.201600626 | |
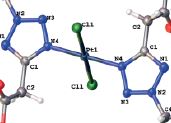 |
Abstract. 2-R-2H-Tetrazol-5-ylacetic acids (abbreviated as 2-Rtaa;R = Me, iPr, tBu) react with K2[PtCl4] in 1 M HCl in H2O at r.t. furnishing trans-platinum(II) complexes trans-[PtCl2(2-R-taa)2] (1–3), whereas cis-isomeric species cis-[PtCl2(2-R-taa)2] (R = iPr, 4; tBu, 5) are isolated at lower temperature (4–6 °C). In the presence of EtOH in the reaction mixture, esterification of the tetrazol-5-ylacetoxy group of 2-tBu-taa leads to trans-[PtCl2(ethyl 2-tert-butyl-2H-tetrazol-5-ylacetate)2] (6). Complexes 1–6 were characterized by elemental analyses (CHN), HRESI+-MS, 1H, 13C{1H}, 195Pt{1H} NMR and IR spectroscopy, differential scanning calorimetry/thermogravimetry (DSC/TG), and X-ray diffraction (for 1·H2O, 2, 3·2H2O, 4, 5·2H2O, and 6). The generation of the tetrazole-based complexes in solution (1 M DCl in D2O, 25 °C) was studied by 1H NMR spectroscopy and HPLCMS. The obtained data indicate the initial formation of anionic [PtCl3(2-R-taa)]– complexes that are subsequently converted into disubstituted isomeric platinum(II) species cis- and trans-[PtCl2(2-R-taa)2]. By contrast to cis- and trans-[PtCl2(2-R-taa)2] that were inactive in two human cancer models in vitro (IC50 > 100 μM), complex 6 demonstrated noticeable antiproliferative effects in HT-29 colon and MCF-7 breast carcinoma cell lines with IC50 values of 14.2 ± 1.1 and 5.8 ± 1.2 μM, respectively. | |
| 29 | Maia N. Putintseva, Olga Yu. Bakulina, Alexander Yu. Ivanov, Pavel S. Lobanov, Sofia K. Nikolskaya, Ilya E. Kolesnikov, Dmitry V. Dar’in “Double tandem cyclization of 4-(1-acyl-2,2-diaminovinyl)-6-arylpyrimidine-5-carbonitriles. Synthesis of novel peri-annulated azines”, Tetrahedron Letters 57 (2016) 5192–5196 DOI: 10.1016/j.tetlet.2016.10.020 | |
 |
Abstract. A series of novel peri-fused azines, pyrimido[4,5,6-de]quinolino[2,3-b][1,8]naphthyridines and benzo[b] pyrimido[4,5,6-de][1,8]naphthyridines, were prepared via the base-promoted double tandem cyclization of 4-(1-acyl-2,2-diaminovinyl)-6-(haloaryl)pyrimidine-5-carbonitriles. This transformation represents a rare example of the one-pot synthesis of peri-annulated polycyclic structures from non-fused heterocycles. The synthesized pyrimido[4,5,6-e]quinolino[2,3-b][1,8]naphthyridines were found to be luminophores. | |
| 30 | Maria A. Sandzhieva, Anna N. Kazakova, Irina A. Boyarskaya, Alexandr Yu. Ivanov, Valentine G. Nenajdenko, and Aleksander V. Vasilyev “Friedel−Crafts Alkylation of Arenes with 2‑Halogeno-2-CF3‑styrenes under Superacidic Conditions. Access to Trifluoromethylated Ethanes and Ethenes”, J. Org. Chem. 2016, 81, 5032−5045 DOI: 10.1021/acs.joc.6b00419 | |
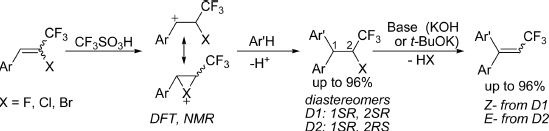 |
Abstract. The formation of the corresponding benzyl cations [ArHC+−CH(X)CF3] takes place under protonation of E-/Z-2-halogeno-2-CF3 styrenes [ArCHC(X)CF3, X = F, Cl, Br] in superacids. The structures of these new electrophiles were studied by means of NMR and theoretical DFT calculations. According to these data, in the case of bromo derivatives, the formed cations, most probably, exist as cyclic bromonium ions; however, in the cases of chloro and fluoro derivatives, open forms are more preferable. Subsequent reaction of these benzyl cations with arenes proceeds as Friedel−Crafts alkylation to afford 1,1-diaryl-2-halo-3,3,3-trifluoropropanes [Ar(Ar′)CH−CH(X)CF3] in high yields (up to 96%) as a mixture of two diastereomers. The prepared halogenopropanes were easily converted into the corresponding mixtures of E-/Z-trifluoromethylated diarylethenes [Ar(Ar′)CCCF3] (in yields up to 96%) by dehydrohalogenation with base (KOH or t-BuOK). The mechanism of elimination (E2 and Ecb) depends on the nature of the leaving group and reaction conditions. |
|
| 31 | Steve Saulnier, Alexander A. Golovanov, Alexandr Yu. Ivanov, Irina A. Boyarskaya,and Aleksander V. Vasilyev “Transformations of Conjugated Enynones in the Superacid CF3SO3H. Synthesis of Butadienyl Triflates, Indanones, and Indenes”, J. Org. Chem. 2016, 81, 1967−1980 DOI: 10.1021/acs.joc.5b02785 | |
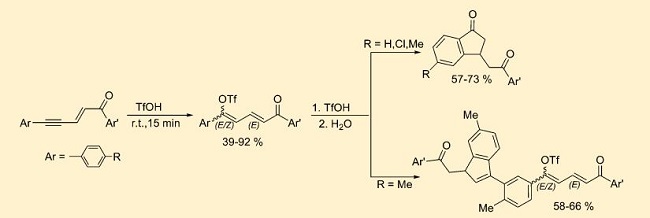 |
Abstract. Conjugated 1,5-diarylpent-2-en-4-yn-1-ones add the superacid CF3SO3H to the acetylenic bond with formation of the corresponding butadienyl triflates. Under superacidic reaction conditions, these triflates are transformed into indanone or indene derivatives depending on which substituents on the aromatic ring are conjugated with the butadiene fragment. In a less acidic system (10% vol pyridine in CF3SO3H) only the formation of butadienyl triflates takes place. Cationic reaction intermediates were studied by means of NMR and DFT calculations. | |
| 32 | Aleksandr N. Shestakov and Mikhail A. Kuznetsov “Synthesis of di-, tri- and tetracyclopropylhydrazines”, Chem. Commun., 2016, 52, 2398-2400 DOI: 10.1039/c5cc07477k | |
| Abstract. Previously unknown 1,1-dicyclopropylhydrazine was obtained in two steps starting from dicyclopropylamine. It serves as a convenient starting material to tri- and tetracyclopropylhydrazines, which have not been described in the literature either. Tricyclopropylhydrazine was prepared in an overall four-step sequence featuring the de Meijere–Chaplinski modification of the Kulinkovich reaction as a key step. Tetracyclopropylhydrazine was obtained by the reductive amination of the cyclopropanone ethyl trimethylsilyl acetal with 1,1-dicyclopropylhydrazine or with the parent hydrazine. Synthetic utility of these cyclopropylhydrazine building blocks is presented as well. |
||
| 33 | Anna A. Shmyreva, Majid Safdari, István Furó, and Sergey V. Dvinskikh “NMR longitudinal relaxation enhancement in metal halides by heteronuclear polarization exchange during magic-angle spinning”, J. Chem. Phys. 144 (2016), 224201 DOI: 10.1063/1.4953540 | |
 |
Abstract. Orders of magnitude decrease of 207Pb and 199Hg NMR longitudinal relaxation times T1 upon magic-angle-spinning (MAS) are observed and systematically investigated in solid lead and mercury halides MeX2 (Me = Pb, Hg and X = Cl, Br, I). In lead() halides, the most dramatic decrease of T1 relative to that in a static sample is in PbI2, while it is smaller but still significant in PbBr2, and not detectable in PbCl2. The effect is magnetic-field dependent but independent of the spinning speed in the range 200–15 000 Hz. The observed relaxation enhancement is explained by laboratory-frame heteronuclear polarization exchange due to crossing between energy levels of spin-1/2 metal nuclei and adjacent quadrupolar-spin halogen nuclei. The enhancement effect is also present in lead-containing organometal halide perovskites. Our results demonstrate that in affected samples, it is the relaxation data recorded under non-spinning conditions that characterize the local properties at the metal sites. A practical advantage of fast relaxation at slow MAS is that spectral shapes with orientational chemical shift anisotropy information well retained can be acquired within a shorter experimental time. | |
| 34 | Mark V. Sigalov, Svetlana A. Pylaeva, and Peter M. Tolstoy “Hydrogen Bonding in Bis(6-amino-1,3-dimethyluracil-5-yl)-methane Derivatives: Dynamic NMR and DFT Evaluation”, J. Phys. Chem. A 2016, 120, 2737−2748 DOI: 10.1021/acs.jpca.6b02184 | |
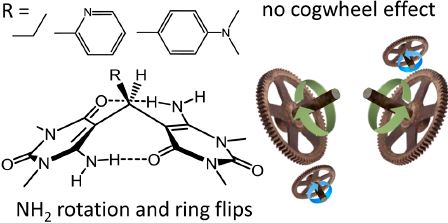 |
Abstract. Three bis(6-amino-1,3-dimethyluracil-5-yl)-methane derivatives were studied experimentally by variabletemperature 1H NMR in polar aprotic solutions (CD2Cl2, C5D5N, C2D2Cl4) and computationally by DFT. The unusual for diarylmethanes coplanar conformation of dimethyluracil rings of each molecule is held by a pair of unequal intramolecular N−H···O hydrogen bonds. We show the presence of two dynamic processes involving breakage/ formation of these bonds. First, it is two independent NH2 group rotations, each coupled to nitrogen inversion. Second, it is uracil ring rotations (ring flips). The thermodynamic parameters (ΔH‡, ΔS‡, and ΔG‡) of both processes were estimated by the full line shape analysis of NMR signals and also by DFT calculations. We demonstrate that, though the ring flips exchange pairs of NH protons, the two processes are not coupled: during the ring flip NH2 groups do not rotate, and during the NH2 rotation the rings do not necessarily rotate. Unlike in many other diarylmethanes, the ring flips in the studied compounds are happening stepwise; i.e., the configuration when both rings are “in flight” at the same time is energetically unfavorable (small degree of “cog wheel effect”). The signs of the ΔS‡ values indicate that the molecular flexibility increases during the NH2 rotations, but decreases during the ring flips. | |
| 35 | Vasily V. Sivchik, Elena V. Grachova, Alexei S. Melnikov, Sergey N. Smirnov, Alexander Yu. Ivanov, Pipsa Hirva, Sergey P. Tunik, and Igor. O. Koshevoy “Solid-State and Solution Metallophilic Aggregation of a Cationic [Pt(NCN)L]+ Cyclometalated Complex”,Inorg. Chem. 2016 DOI: 10.1021/acs.inorgchem.5b02713 | |
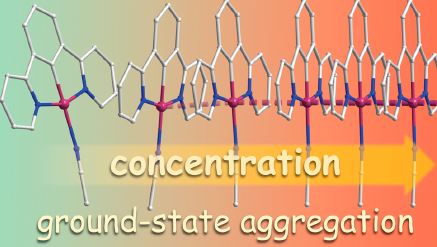 |
Abstract. The noncovalent intermolecular interactions (π−π stacking, metallophilic bonding) of the cyclometalated complexes [Pt(NCN)L]+X− (NCN = dipyridylbenzene, L = pyridine (1), acetonitrile (2)) are determined by the steric properties of the ancillary ligands L in the solid state and in solution, while the nature of the counterion X− (X− = PF6−, ClO4−, CF3SO3−) affects the molecular arrangement of 2·X in the crystal medium. According to the variable-temperature Xray diffraction measurements, the extensive Pt···Pt interactions and π-stacking in 2·X are significantly temperature-dependent. The variable concentration 1H and diffusion coefficients NMR measurements reveal that 2·X exists in the monomeric form in dilute solutions at 298 K, while upon increase in concentration [Pt(NCN)(NCMe)]+ cations undergo the formation of the ground-state oligomeric aggregates with an average aggregation number of ∼3. The photoluminescent characteristics of 1 and 2·X are largely determined by the intermolecular aggregation. For the discrete molecules the emission properties are assigned to metal perturbed IL charge transfer mixed with some MLCT contribution. In the case of oligomers 2·X the luminescence is significantly red-shifted with respect to 1 and originates mainly from the 3MMLCT excited states. The emission energies depend on the structural arrangement in the crystal and on the complex concentration in solution, variation of which allows for the modulation of the emission color from greenish to deep red. In the solid state the lability of the ligands L leads to vapor-induced reversible transformation 1 ↔ 2 that is accompanied by the molecular reorganization and, consequently, dramatic change of the photophysical properties. Time-dependent density functional theory calculations adequately support the models proposed for the rationalization of the experimental observations. |
|
| 36 | Michael A. Smirnov, Maria P. Sokolova, Natalya V. Bobrova, Igor A. Kasatkin, Erkki Lahderanta, Galina K. Elyashevich “Capacitance properties and structure of electroconducting hydrogels based on copoly(aniline e p-phenylenediamine) and polyacrylamide”,Journal of Power Sources 304 (2016) 102-110 DOI: 10.1016/j.jpowsour.2015.11.035 | |
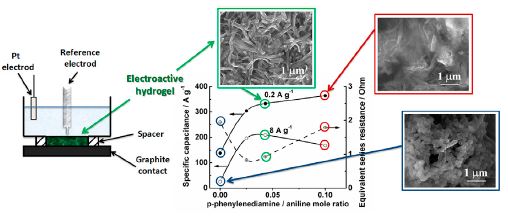 |
Abstract. Electroconducting hydrogels (EH) based on copoly(aniline e p-phenylenediamine) grafted to the polyacrylamide for the application as pseudo-supercapacitor’s electrodes have been prepared. The influence of preparation conditions on the structure and capacitance properties of the systems were investigated: we determined the optimal amount of p-phenylenediamine to obtain the network of swollen interconnected nanofibrils inside the hydrogel which provides the formation of continuous conducting phase. Structure and morphology of the prepared samples were investigated with UVeVIS spectroscopy, scanning electron microscopy (SEM) and wide-angle X-ray diffraction (WAXD). The maximal value of capacitance was 364 F g1 at 0.2 A g1. It was shown that the EH samples demonstrate the retention of 50% of their capacity at high current density 16 A g1. Cycle-life measurements show evidence that capacitance of EH electrodes after 1000 cycles is higher than its initial value for all prepared samples. Changes of the copolymer structure during swelling in water have been studied with WAXD. | |
| 37 | Elena V. Solovyeva, Galina L. Starova, Liubov A. Myund, Anna S. Denisova “X-ray, IR and Raman study of Ag(I), Cu(II) and Cd(II) complexes with 4,5-bis(N,N-di(2-hydroxyethyl)iminomethyl)acridine”, Polyhedron 106 (2016) 1–9 DOI: 10.1016/j.poly.2015.12.049 | |
 |
Abstract. Five new 1:1 metal complexes of highly selective acridine-based fluorophore 4,5-bis(N,N-di(2-hydroxyethyl) iminomethyl)acridine (BHIA) with Ag(I), Cu(II) and Cd(II) were synthesized, characterized and investigated by the methods of single crystal X-ray diffraction, IR, and Raman spectroscopy. Single crystal X-ray analysis reveals that three different coordination manners of the BHIA occur. Five atoms of the BHIA molecule take part in the formation of complex with Ag(I) and six atoms are involved in the formation of complex with Cu(II). In the case of BHIA with Cd(II) interaction, the coordination possibilities of the cation depend on an anion and vary from five to seven. The investigation of BHIA complexes by IR and Raman spectroscopy shows that if there are two free hydroxyl groups in the molecule then the vibrational spectrum arises from many rotational conformations. The different binding manner of BHIA molecule with metal cations is the most clearly manifested in the IR spectrum in the region of the stretching OH-vibrations. | |
| 38 | Mariia V. Sorokina, Alena S. Pankova, and Mikhail A. Kuznetsov “Oxidative Aminoaziridination of 2-Vinylfuran Derivatives as an Approach to Hexa-2,5-diene-1,4-dione Monohydrazones”, Asian J. Org. Chem. 2016, 5, 389 – 398 DOI: 10.1002/ajoc.201500460 | |
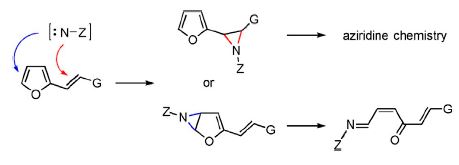 |
Abstract. The oxidative addition of N-aminophthalimide to substituted 2-vinylfurans provides monophthaloylhydrazones of (2Z)-hexa-2,5-diene-1,4-dione derivatives instead of 2-furylaziridines by proceeding through an aziridination of the endocyclic furan C=C bond followed by a regio- and stereoselective rearrangement of the bicyclic intermediate. The formation of stable aziridines can then occur through the aziridination of the (E)-C=C bond of these phthaloylhydrazones. Detailed structure elucidations and mechanistic considerations are provided. | |
| 39 | Nicolay G. Voron’ko, Svetlana R. Derkach, Mikhail A. Vovk, Peter M. Tolstoy “Formation of -carrageenan–gelatin polyelectrolyte complexes studied by1H NMR, UV spectroscopy and kinematic viscosity measurements”, Carbohydrate Polymers 151 (2016) 1152–1161 DOI: 10.1016/j.carbpol.2016.06.060 | |
 |
Abstract. The intermolecular interactions between an anionic polysaccharide from the red algae -carrageenanand a gelatin polypeptide, forming stoichiometric polysaccharide–polypeptide (bio)polyelectrolyte com-plexes in the aqueous phase, were examined. The major method of investigation was high-resolution 1HNMR spectroscopy. Additional data were obtained by UV absorption spectroscopy, light scattering dis-persion and capillary viscometry. Experimental data were interpreted in terms of the changing roles of electrostatic interactions, hydrophobic interactions and hydrogen bonds when -carrageenan–gelatin complexes are formed. At high temperatures, when biopolymer macromolecules in solution are in the state of random coil, hydrophobic interactions make a major contribution to complex stabilization. At the temperature of gelatin’s coil → helix conformational transition and at lower temperatures, electrostatic interactions and hydrogen bonds play a defining role in complex formation. A proposed model of the-carrageenan–gelatin complex is discussed. | |
| 40 | Ekaterina S. Yandanova, Daniil M. Ivanov, Maxim L. Kuznetsov, Andrey G. Starikov, Galina L. Starova, and Vadim Yu. Kukushkin “Recognition of S···Cl Chalcogen Bonding in Metal-Bound Alkylthiocyanates”,Cryst. Growth Des. 2016, 16, 2979−2987 DOI: 10.1021/acs.cgd.6b00346 | |
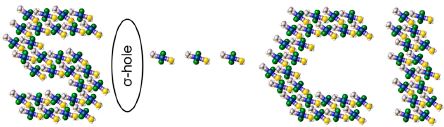 |
Abstract. Reaction of K2[PtCl4] with excess AlkSCN in water gives the alkylthiocyanate complexes trans-[PtCl2(AlkSCN)2] (Alk = Et 1, nPr 2; 80–85%). These species were studied, in particular, by X-ray crystallography. In the solid state, both 1 and 2 exhibit the previously unreported S···Cl chalcogen bonding, which consolidates the complexes into networks and leads to layered structures. Theoretical density functional theory calculations and Bader’s atoms in molecules analysis demonstrated two types of intermolecular interactions in tetramer (1)4, viz. the S···Cl chalcogen and the H···Cl hydrogen bonds. Despite that each particular S···Cl or H···Cl bonding is weak with the estimated energy of 1–2 kcal/mol, altogether they play a crucial role in the stabilization of the S2Cl2 fragment in (1)4, the basis set of superposition error corrected interaction energy being −12.8 kcal/mol per monomer complex molecule. The chalcogen bonding and the rhomboidal structure of the S2Cl2 fragment can be interpreted in terms of electrostatic arguments as a result of the interaction between the belt of negative electrostatic potential around the Cl atoms and the sulfur σ-holes. The natural bond orbital analysis revealed that both LP(S) → LP*(Pt)/σ*(Pt–N)/σ*(Pt–Cl) and LP(Cl) → σ*(S–C) types of hyperconjugative charge transfers are important in the chalcogen bonding. | |
| 41 | А.Ф. Сафитдинова, С.А. Галкина, Е.И. Кошель, Е.Р. Гагинаская “Роль повторяющихся последовательностей в эволюции половых хромосом у птиц”, Цитология том. 58 (№5, 2016) 393-398 | |
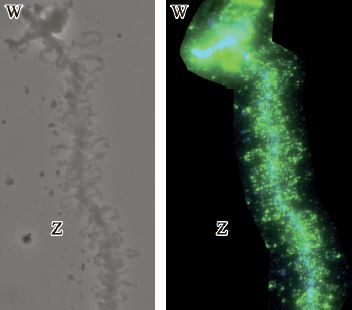 |
Abstract. В настоящей работе мы проанализировали распределение и траскрипционную активность представителей известных семейств повторяющихся элементов в составе половых хромосом у курицы на стадии ламповых щеток. На основе полученных данных мы предположили участие рассеяных повторов в поддержании эволюционно значимого уровня изменчивости гетероморфных половых хромосом у птиц. Исследование особенности организации повторов, специфичных для половой хромосомы W курицы, позволило сформулировать гипотезу о роли накопления обогащенных гомопуриновыми участками тандемных повторов в эволюции половых хромосом. | |
| 42 | Н. В. Шугурова, Е. И. Давыдова, Т. Н. Севастьянова, А. Д. Мишарев, M. Боденштайнер, M. Шеер “ВЗАИМОДЕЙСТВИЕ TiCl4 С ДИЭТИЛОВЫМ ЭФИРОМ. ЭКСПЕРИМЕНТАЛЬНОЕ И КВАНТОВОХИМИЧЕСКОЕ ИССЛЕДОВАНИЕ”, Журнал общей химии Т. 86. Вып. 1 (2016) 11-19 | |
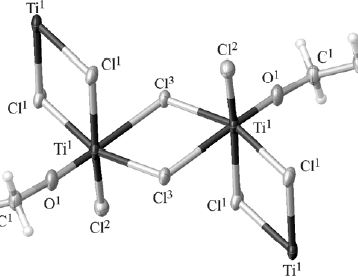 |
Abstract. Методами ЯМР и масс-спектрометрии изучено взаимодействие тетрахлорида титана с диэтиловым эфиром при варьировании соотношения компонентов и температуры. Комплекс TiCl4·2Et2O в газовую фазу не переходит. Образование этокситрихлортитана в продуктах взаимодействия установлено методом рентгеноструктурного анализа. На основании полученных экспериментальных данных и квантовохимических расчетов (метод B3LYP/def2-SVP) предложена модель перехода от исходных компонентов к диоксиду титана. | |
| 43 | A.V. Artem’ev, E.P. Doronina, M.I. Rakhmanova, A.O. Sutyrina, I.Yu. Bagryanskaya, P.M. Tolstoy, A.L. Gushchin, A.S. Mazur, N.K. Gusarova, B.A. Trofimov “Luminescent CuI thiocyanate complexes based on tris(2-pyridyl)phosphine and its oxide: from mono-, di- and trinuclear species to coordination polymer”, New J. Chem. 2016, 40, 10028-10040 DOI:10.1039/C6NJ02087A | |
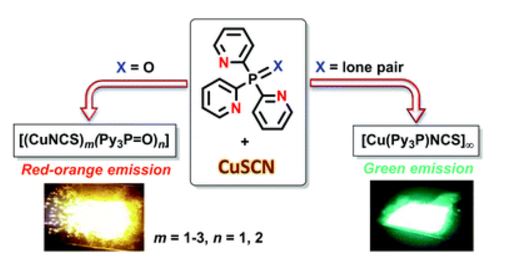 |
Abstract. Tris(2-pyridyl)phosphine oxide reacts with CuSCN to form a variety of luminescent complexes, depending on the specified metal-to-ligand ratio and the solvent used, viz. mononuclear [Cu(N,N′,N′′-Py3P=O)(NCS)], dinuclear (N,N′-Py3P=O)Cu(SCN NCS)Cu[(N,N′-Py3P=O)], their co-crystal (2 : 1, correspondingly) and trinuclear {Cu(NCS)[SCNCu(N,N′,N′′-Py3P=O)]2}. In the solid state, these complexes feature red-orange emission upon UV photoexcitation. The reaction of tris(2-pyridyl)phosphine with CuSCN quantitatively produces an almost insoluble coordination polymer, [Cu(Py3P)NCS]n, which exhibits bright green emission. The synthesized compounds are the first members of the hitherto unknown family of Cu(I) thiocyanate complexes supported by tripodal ligands. | |
| 44 | A.S. Pankova, P.R. Golubev, A.F. Khlebnikov, A.Yu. Ivanov, M.A. Kuznetsov “Thiazol-4-one derivatives from the reaction of monosubstituted thioureas with maleimides: structures and factors determining the selectivity and tautomeric equilibrium in solution”, Beilstein J. Org. Chem., 2016, 12, 2563-2569 DOI:10.3762/bjoc.12.251 | |
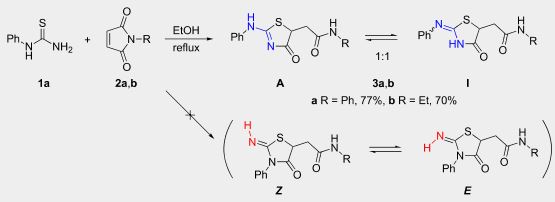 |
Abstract. 2-(Alkyl(aryl)amino)thiazol-4(5H)-ones can regioselectively be prepared from monoalkyl(aryl)thioureas and maleimides. In solution, the former heterocycles exist in a tautomeric equilibrium with 2-(alkyl(aryl)imino)thiazolidin-4-ones and the substituent on the exocyclic nitrogen atom governs the ratio of these tautomers. Isomers with the alkyl group in the endocyclic position can be obtained from N-methyl(ethyl)thioureas. 2D NMR spectroscopy and DFT calculations rationalize experimental results. | |
| 45 | P. Golubev, E.A. Karpova, A.S. Pankova, M. Sorokina, M.A. Kuznetsov “Regioselective Synthesis of 7‑(Trimethylsilylethynyl) pyrazolo [1,5‑a] pyrimidines via Reaction of Pyrazolamines with Enynones”, J. Org. Chem., 2016, 81, 11268-11275 DOI:10.1021/acs.joc.6b02217 | |
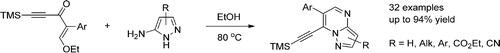 |
Abstract. Condensation of enynones readily available from cheap starting material with pyrazolamines provides easy access to fluorescent 7-(trimethylsilylethynyl)pyrazolo[1,5-a]pyrimidines. The reaction is straightforward, does not require the use of any additional reagents or catalysts, and can be performed without inert atmosphere. Various substituents and functional groups in both enynone and pyrazolamine are tolerated. The presented method features full regioselectivity, high isolated yields, and simplicity of both setup and product purification. Fluorescent properties of the obtained pyrazolopyrimidines were studied. | |
| 46 | B. Koeppe, S.A. Pylaeva, C. Allolio, D. Sebastiani, E.T.J. Nibbering, G.S. Denisov, H.-H. Limbach, P.M. Tolstoy “Polar solvent fluctuations drive proton transfer in hydrogen bonded complexes of carboxylic acid with pyridines: NMR, IR and ab initio MD study”, Phys. Chem. Chem. Phys., 2016, accepted DOI:10.1039/C6CP06677A | |
 |
Abstract. We study a series of intermolecular hydrogen-bonded 1 : 1 complexes formed by chloroacetic acid with 19 substituted pyridines and one aliphatic amine dissolved in CD2Cl2 at low temperature by 1H and 13C NMR and FTIR spectroscopy. The hydrogen bond geometries in these complexes vary from molecular (O–H…N) to zwitterionic (O…H–N+) ones, while NMR spectra show the formation of short strong hydrogen bonds in intermediate cases. Analysis of CQO stretching and asymmetric CO2 stretching bands in FTIR spectra reveal the presence of proton tautomerism. On the basis of these data, we construct the overall proton transfer pathway. In addition to that, we also study by use of ab initio molecular dynamics the complex formed by chloroacetic acid with 2-methylpyridine, surrounded by 71 CD2Cl2 molecules, revealing a dualmaximum distribution of hydrogen bond geometries in solution. The analysis of the calculated trajectory shows that the proton jumps between molecular and zwitterionic forms are indeed driven by dipole–dipole solvent–solute interactions, but the primary cause of the jumps is the formation/breaking of weak CH…O bonds from solvent molecules to oxygen atoms of the carboxylate group. | |
| 47 | L. Sobczyk, D. Chudoba, P.M. Tolstoy, A. Filarowski “Some Brief Notes on Theoretical and Experimental Investigations of Intramolecular Hydrogen Bonding”, Molecules, 2016, 21, 1657-1/19 DOI:10.3390/molecules21121657 | |
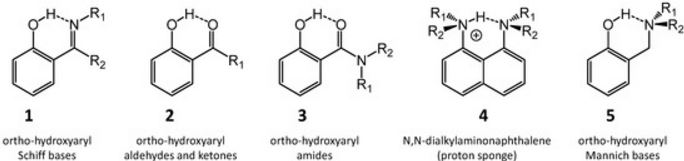 |
Abstract. A review of selected literature data related to intramolecular hydrogen bonding in ortho-hydroxyaryl Schiff bases, ortho-hydroxyaryl ketones, ortho-hydroxyaryl amides, proton sponges and ortho-hydroxyaryl Mannich bases is presented. The paper reports on the application of experimental spectroscopic measurements (IR and NMR) and quantum-mechanical calculations for investigations of the proton transfer processes, the potential energy curves, tautomeric equilibrium, aromaticity etc. Finally, the equilibrium between the intra- and inter-molecular hydrogen bonds in amides is discussed. | |
| 48 | M.A. Kuznetsov, A.N. Shestakov, M. Zibinsky, M. Krasavin, C.T. Supuran, S. Kalinin, M. Tanç “Synthesis, structure and properties of N-aminosaccharin – A selective inhibitor of human carbonic anhydrase I”, Tetrahedron Letters, 2017, 58, 172–174 DOI:10.1016/j.tetlet.2016.12.005 | |
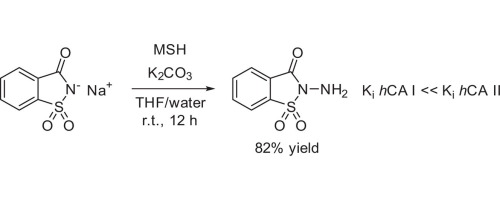 |
Abstract. Previously unknown N-aminosaccharin was prepared in good yield via the one-step direct amination of saccharin sodium salt with hydroxylamine-O-mesitylenesulfonic acid (MSH) and its reactivity investigated. N-aminosaccharin and its derivatives were tested against hCA isoforms and the parent compound was identified to be a selective, low micromolar inhibitor (Ki = 8.8 lM) of hCA I. These findings provide a ligand-efficient starting point for the design of potent hCA I inhibitors – a promising drug target for retinal/cerebral edema treatment. | |
| 49 | Y.M. Zhukov, A.N. Kovalyov, A.Y. Kultaeva, M.G. Shelyapina, «A comparative analysis of the protonated and copper exchanged mordenites with SiO2/Al2O3 molar ratio equal to 10», Int. J. Nanotechnol. 2016, 13, 136-146 DOI:10.1504/IJNT.2016.074529 | |
 |
Abstract.Ion-exchange zeolites are the starting materials for the synthesis of nanocomposites. Moreover, they are of great interest in themselves, because cations in zeolites, and especially cations of transition metals, are the active catalytic sites. Copper-exchange zeolites are known as very effective deNOx catalysts. Although these compounds have been extensively studied in the last decade, the localisation of the copper ions in the zeolite matrix and their state are not fully determined. Here, we report on the results of our complementary study by various experimental methods, such as thermal analysis, nuclear magnetic resonance (NMR), electronic paramagnetic resonance (EPR), and theoretical density functional calculations of protonated and copper-exchanged mordenites with the molar SiO2/Al2O3 ratio equal to 10 | |
| 50 | A.A. Belyaev, D.V. Krupenya, E.V. Grachova, V.V. Gurzhiy, A.S. Melnikov, P.Yu. Serdobintsev, E.S. Sinitsyna, E.G. Vlakh, T.B. Tennikova, S.P. Tunik, «Supramolecular AuI–CuI Complexes as New Luminescent Labels for Covalent Bioconjugation», Bioconjugate Chem. 2016, 27, 143-150 DOI:10.1021/acs.bioconjchem.5b00563 | |
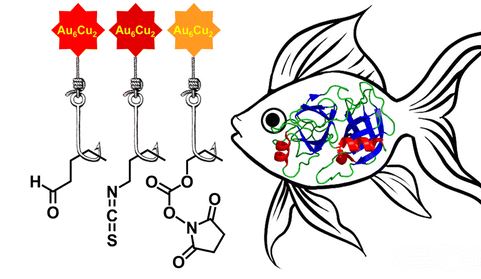 |
Abstract.Two new supramolecular organometallic complexes, namely, [Au6Cu2(C2C6H4CHO)6(PPh2C6H4PPh2)3](PF6)2 and [Au6Cu2(C2C6H4NCS)6(PPh2C6H4PPh2)3](PF6)2, with highly reactive aldehyde and isothiocyanate groups have been synthesized and characterized using X-ray crystallography, ESI mass spectrometry, and NMR spectroscopy. The compounds obtained demonstrated bright emission in solution with the excited-state lifetime in microsecond domain both under single- and two-photon excitation. The luminescent complexes were found to be suitable for bioconjugation in aqueous media. In particular, they are able to form the covalent conjugates with proteins of different molecular size (soybean trypsin inhibitor, human serum albumin, rabbit anti-HSA antibodies). The conjugates demonstrated a high level of the phosphorescent emission from the covalently bound label, excellent solubility, and high stability in physiological media. The highest quantum yield, storage stability, and luminance were detected for bioconjugates formed by covalent attachment of the aldehyde-bearing supramolecular AuI–CuI complex. The measured biological activity of one of the labeled model proteins clearly showed that introduced label did not prevent the biorecognition and specific protein–protein complex formation that was extremely important for the application of the conjugates in biomolecular detection and imaging. | |
| 51 | D.V. Semenok, J.J. Medvedev, M.S. Avdontceva, S.I. Selivanov, J. Sieler, A.S. Mereshchenko, V.A. Nikolaev, “Experimental Evidence of Intramolecular CAr–H•••O=C Hydrogen Bonds in the Structure of (Diaryl)tetrahydrofuranones Using Spectroscopic Tools”, Helvetica Chim. Acta 2016, 99, 716-723 DOI:10.1002/hlca.201600149 | |
 |
Abstract. The occurrence of bifurcate H-bonds CAr–H···O=C in the structure of (diaryl)-tetrahydrofuranones was experimentally demonstrated using different methods and techniques. The consistent increasing spin–spin coupling constants 1J(C,H) of the ortho-H-atoms and low-field shift of vC=O in IR spectra of 2,2-(diaryl)tetrahydrofuran-3(2H)-ones relative to their 5,5-diaryl counterparts, as well as pronounced dependence of the ortho-C–H H-atoms chemical shifts on the temperature and solvent polarity along with X-ray diffraction analysis data unambiguously point to the existence of weak CAr–H···O=C H-bonds in these molecules. | |
| 52 | V.A. Rassadin, D.P. Zimin, G.Z. Raskil’dina, A.Yu. Ivanov, V.P. Boyarskiy, S.S. Zlotskii, V.Yu. Kukushkin, “Solvent- and halide-free synthesis of pyridine-2-yl substituted ureas through facile C–H functionalization of pyridine N-oxides”, Green Chem. 2016, Advance Article DOI:10.1039/C6GC02556K | |
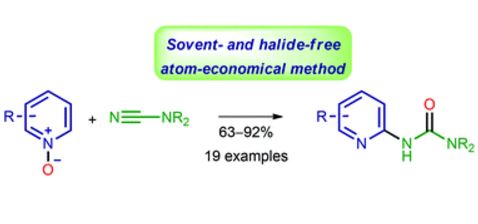 |
Abstract. The occurrence of bifurcate H-bonds CAr–H···O=C in the structure of (diaryl)-tetrahydrofuranones was experimentally demonstrated using different methods and techniques. The consistent increasing spin–spin coupling constants 1J(C,H) of the ortho-H-atoms and low-field shift of vC=O in IR spectra of 2,2-(diaryl)tetrahydrofuran-3(2H)-ones relative to their 5,5-diaryl counterparts, as well as pronounced dependence of the ortho-C–H H-atoms chemical shifts on the temperature and solvent polarity along with X-ray diffraction analysis data unambiguously point to the existence of weak CAr–H···O=C H-bonds in these molecules. | |
| 53 | A.M. Afanasenko, M.S. Avdontceva, A.S. Novikov, T.G. Chulkova, “Halogen and hydrogen bonding in cis- dichlorobis (propionitrile) platinum(II) chloroform monosolvate”, Z. Kristallogr. Cryst. Mater. 2016, 231, 435-440 DOI:10.1515/zkri-2016-1948 | |
 |
Abstract. The weak intermolecular noncovalent interactions in the solvate cis-[PtCl2(C2H5CN)2]·CHCl3 were studied by XRD and DFT methods. Special attention was paid to a chloroform molecule as a potential donor of both hydrogen and halogen bonds. On the one hand, the H atom of chloroform links two Cl atoms of cis-[PtCl2(C2H5CN)2] (Hchloroform–Clcomplex (2.660 Å, 2.811 Å)), and on the other side, the weak Clchloroform–Clcomplex (3.247 and 3.497 Å) interactions were found. According to DFT calculation, only the Hchloroform–Clcomplex (2.660 and 2.811 Å) and Clchloroform–Clcomplex (3.247 Å) interactions can be classified as weak noncovalent bonds, whereas the contacts Clchloroform–Cnitrile (3.391 Å) and Clchloroform–Clcomplex (3.497 Å) are due to the crystal packing effect | |
| 54 | E.V. Alopina, E.A. Safonova, I.B. Pukinsky, N.A. Smirnova, “Liquid−Liquid Equilibria in Aqueous Mixtures of Alkylmethylimidazolium Glutamate with Potassium Carbonate and Some Physicochemical Properties of Aqueous [Cnmim][Glu] (n = 4, 6, 8) Solutions”, J. Chem. Eng. Data 2016, 61, 2013-2019 DOI:10.1021/acs.jced.5b00948 | |
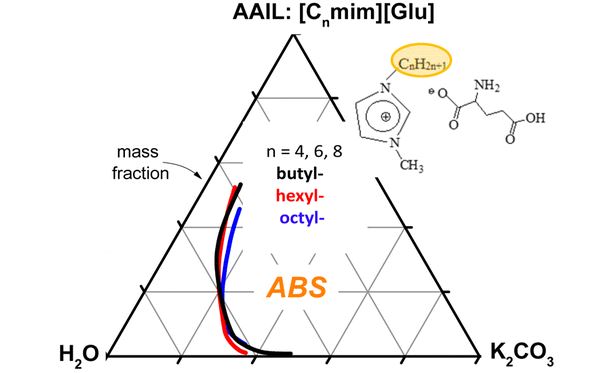 |
Abstract. Aqueous biphasic systems containing ionic liquids (ILs) attract rather high attention; in particular their application in liquid–liquid extraction is very attractive due to the specificity of the systems and great possibilities in the modification of IL chemical structures. In the present work, a series of amino acid ionic liquids (AAILs) based on the 1-alkyl-3-methylimidazolium cation and l-glutamic acid anion [Cnmim][Glu] (n = 4, 6, 8) were synthesized. Densities, refractive indices, and specific electrical conductivities of aqueous solutions of these AAILs in the concentration range 0–70 wt % (for [C4mim][Glu] solutions up to 87 wt %) were measured at temperature 298.15 K and ambient pressure 0.1 MPa. The liquid–liquid equilibria for ternary solutions [Cnmim][Glu] + salt (K2CO3) + water at 298.15 K were under study. In aqueous solutions of [C8mim][Glu] micellization is observed, whereas [C4mim][Glu] and [C6mim][Glu] in aqueous solutions form small ordinary aggregates. The critical micellar concentration for [C8mim][Glu] aqueous solutions at 298.15 K was estimated from the experimental data on the concentration dependence of the specific electrical conductivities of the solutions. | |
| 55 | S.N. Britvin, A.M. Rumyantsev, A.E. Zobnina, M.V. Padkina, “Between Adamantane and Atrane: Intrabridgehead Interactions in the Cage-Like Phosphane Related to a Novel Tris(homoadamantane) Ring System”, Chem. Eur. J. 2016, 22, 14227-14235 DOI:10.1002/chem.201601637 | |
 |
Abstract. Herein, we report unusual four-center interactions in the novel cage-like phosphane, 1,4,7-triaza-9-phosphatricyclo[5.3.2.14,9]tridecane (CAP). This water-soluble ligand, the first example of a tris(homoadamantane) ring system, can be considered a macrocyclic homologue of the well-known PTA (1,3,5-triaza-7-phosphaadamantane). However, 31P NMR spectroscopic anomalies of CAP follow those typical for the bi-/tricyclic atrane systems. Another atrane-like feature of CAP is the ability of one nitrogen atom to undergo out–in pyramidal inversion. The latter is associated with a substantial decrease in the intracage N−N and P−N distances. Analysis of electron density distribution [molecular electrostatic potential (MESP) and atoms-in-molecules (AIM) approaches] suggests that the P and N atoms in the pyramidally inverted CAP derivatives are involved in interactions resulting in accumulation of electron density at the center of the phosphane cage. The latter can reliably explain the stereoelectronic and NMR anomalies of the new ligand. The semi-flexible CAP cage populates the structural niche between the rigid adamantine skeleton of PTA and flexible atrane systems and can be regarded as an alternative to PTA in aqueous coordination chemistry. | |
| 56 | S.N. Britvin, A.M. Rumyantsev, A.E. Zobnina, M.V. Padkina, “Molecular structure, interatomic interactions and vibrational analysis of 1,4-diazabicyclo[3.2.1]octane parent ring system”, J. Mol. Struct. 2016, 1130, 395-399 DOI:10.1016/j.molstruc.2016.10.065 | |
 |
Abstract. Molecular structure of 1,4-diazabicyclo[3.2.1]octane, a parent ring of TAN1251 family of alkaloids, is herein characterized for the first time in comparison with the structure of nortropane (8-azabicyclo[3.2.1]octane), the parent framework of tropane ring system. The methods of study involve X-ray structural analysis, DFT geometry optimizations with infrared frequency calculations followed by natural bond orbital (NBO) analysis, and vibrational analysis of infrared spectrum. | |
| 57 | M. Chizhova, O. Bakulina, D. Dar’in, M. Krasavin, “New Dicarboxylic Acid Anhydride for Ambient-Temperature Castagnoli-Cushman Reactions”, ChemistrySelect 2016, 1, 5487-5492 DOI:10.1002/slct.201601207 | |
 |
Abstract. A novel 3-phenyl-4-thiaglutaric anhydride was employed as a highly reactive partner for the Castagnoli-Cushman reaction. This, in combination with the use of DMF as a reaction medium, resulted in a drastic lowering of the reaction temperature to ambient (compared to the 110–150 °C range typical for similar processes involving glutaric anhydride) and an extended reaction scope (with respect to substitution pattern in the imine component). Diastereomeric products were separated, in the form of respective methyl esters, and their comparative NMR characterization led to preliminary generalizations regarding the syn- and anti-series. | |
| 58 | D. Dar’in, O. Bakulina, S. Nikolskaya, I. Gluzdikov, M. Krasavin, “The rare cis-configured trisubstituted lactam products obtained by the Castagnoli–Cushman reaction in N,N-dimethylformamide”, RSC Adv. 2016, 6, 49411-49415 DOI:10.1039/c6ra10249b | |
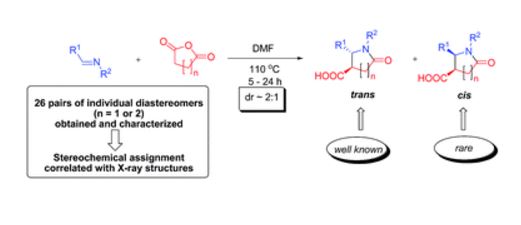 |
Abstract. Unlike its trans counterpart, the cis-configured scaffold derived from the Castagnoli–Cushman reaction (CCR) is scarce and has not been explored in bioactive compound design. We found that conducting this reaction in DMF (in contrast to conventional toluene or xylene) led to a significantly higher proportion of cis-configured lactams in the diastereomeric product mixture. This allowed us, for the first time, to obtain and thoroughly characterize both stereoisomers of a significant number of lead-like CCR lactams. Simple rules for 1H NMR-based stereochemical assignment have been devised and correlated with the single-crystal X-ray structures obtained for pure cis- and trans-configured lactams. | |
| 59 | M.M. Efremova, A.P. Molchanov, A.G. Larina, G.L. Starova, R.R. Kostikov, A.V. Stepakov, “Acid-Induced Rearrangement of Cycloadducts from Cyclopropenecarboxylates and 1,3-Diarylisobenzofurans”, Helv. Chim. Acta 2016, 99, 487-493 DOI:10.1002/hlca.201500114 | |
 |
Abstract. Treatment of several Diels–Alder adducts of cyclopropenecarboxylates and 1,3-diarylisobenzofurans with a strong acid triggers a skeletal rearrangement resulting in 4,8b-dihydro-3aH-indeno[1,2-b]furans. | |
| 60 | M.R. Galding, A.V. Virovets, I.V. Kazakov, M. Scheer, S.N. Smirnov, A.Y. Timoshkin, “Diminished electron density in the Vaska-type rhodium(I) complex trans-[Rh (NCBH3) (CO)(PPh3)2]”, Acta Cryst. 2016, C72, 514-517 DOI:10.1107/S2053229616008536 | |
 |
Abstract. Vaska-type complexes, i.e. trans-[RhX(CO)(PPh3)2] (X is a halogen or pseudohalogen), undergo a range of reactions and exhibit considerable catalytic activity. The electron density on the RhI atom in these complexes plays an important role in their reactivity. Many cyanotrihydridoborate (BH3CN-) complexes of Group 6-8 transition metals have been synthesized and structurally characterized, an exception being the rhodium(I) complex. Carbonyl(cyanotrihydridoborato-[kappa]N)bis(triphenylphosphine-[kappa]P)rhodium(I), [Rh(NCBH3)(CO)(C18H15P)2], was prepared by the metathesis reaction of sodium cyanotrihydridoborate with trans-[RhCl(CO)(PPh3)2], and was characterized by single-crystal X-ray diffraction analysis and IR, 1H, 13C and 11B NMR spectroscopy. The X-ray diffraction data indicate that the cyanotrihydridoborate ligand coordinates to the RhI atom through the N atom in a trans position with respect to the carbonyl ligand; this was also confirmed by the IR and NMR data. The carbonyl stretching frequency [nu](CO) and the carbonyl carbon 1JC-Rh and 1JC-P coupling constants of the Cipso atoms of the triphenylphosphine groups reflect the diminished electron density on the central RhI atom compared to the parent trans-[RhCl(CO)(PPh3)2] complex. | |
| 61 | P. Golubev, O. Bakulina, D. Dar’in, M. Krasavin, “Indoline-Based Constrained Peptidomimetic Motifs Obtained via the Joullié–Ugi Reaction of Indolenines”,Eur. J. Org. Chem. 2016, 3969-3976 DOI:10.1002/ejoc.201600656 | |
 |
Abstract. Sterically demanding indolenines were employed as cyclic imine substrates in the Joullié–Ugi reaction with diverse isocyanides and carboxylic acids resulting in a new type of conformationally constrained indoline-based peptidomimetic compounds. | |
| 62 | L.Yu. Gurskaya, D.S. Belyanskaya, D.S. Ryabukhin, D.I. Nilov, I.A. Boyarskaya, A.V. Vasilyev, “Reactions of N,3-diarylpropiolamides with arenes under superelectrophilic activation: synthesis of 4,4-diaryl-3,4-dihydroquinolin-2-(1H)-ones and their derivatives”,Beilstein J. Org. Chem. 2016, 12, 950-956 DOI:10.3762/bjoc.12.93 | |
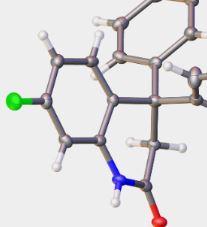 |
Abstract. The reaction of 3-aryl-N-(aryl)propiolamides with arenes in TfOH at room temperature for 0.5 h led to 4,4-diaryl-3,4-dihydroquinolin-2-(1H)-ones in yields of 44–98%. The obtained dihydroquinolinones were further transformed into the corresponding N-acyl or N-formyl derivatives. For the latter, the superelectrophilic activation of the N-formyl group by TfOH in the reaction with benzene resulted in the formation of N-(diphenylmethyl)-substituted dihydroquinolinones. | |
| 63 | A.N. Kazakova, C.G. Nenajdenko, A.V. Vasilyev, “Oxidative Dimerization of 2-Chloro-3,3,3-trifluoro-1-(4-methoxyphenyl)propene in the System PbO2–CF3SO3H”, Russ. J. Org. Chem. 2016, 52, 594-595 DOI:10.1134/S1070428016040230 | |
| 64 | A.A. Kazakova, A.A. Bogomazova, R.O. Iakovenko, S.S. Zlotsky, A.V. Vasilyev, “TfOH promoted reactions of vinyl gem-dichlorocyclopropanes with arenes: access to aryl gem-dichloropentenes”, Tetrahedron Lett. 2016, 56, 4210-4212 DOI:10.1016/j.tetlet.2016.08.008 | |
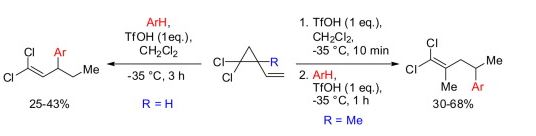 |
Abstract. The reaction of 1,1-dichloro-2-vinylcyclopropane with arenes under the action of the superacid TfOH afforded 3-aryl-1,1-dichloropent-1-enes (25–43% yield), as the products of cyclopropane ring opening. The reaction of 1,1-dichloro-2-methyl-2-vinylcyclopropane was realized as a two-step procedure, including initial in situ generation of the corresponding triflate, 5,5-dichloro-4-methylpent-4-en-2-yl trifluoromethanesulfonate, from the cyclopropane and TfOH, followed by reaction with arenes and TfOH to give 1,1-dichloro-2-methyl-4-arylpent-1-enes (30–68% yield). | |
| 65 | M.A. Klingenberg, A.S. Bogachenkov, M.A. Kinzhalov, A.V. Vasilyev, V.P. Boyarskiy, “1,4-Dihydrophosphinolines and their complexes with group 10 metals”, New J. Chem. 2016, 40, 3336-3342 DOI:10.1039/c5nj03038b | |
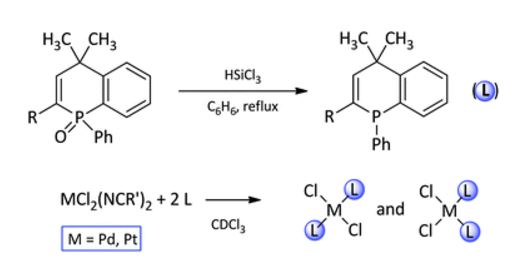 |
Abstract. Syntheses and characterizations of novel phosphaheterocycles, 1-phenyl-4,4-dimethyl-1,4-dihydrophosphinoline (2a) and 1-phenyl-2-bromo-4,4-dimethyl-1,4-dihydrophosphinoline (2b), and their complexes with Pd(II) and Pt(II) are described. Reduction of 1-phenyl-4,4-dimethyl-1,4-dihydrophosphinoline 1-oxide (1a) and 1-phenyl-2-bromo-4,4-dimethyl-1,4-dihydrophosphinoline 1-oxide (1b) using trichlorosilane in refluxing benzene gave the target phosphines in a yield of 83–87%. Reactions of phosphines with acetonitrile complexes of Pd(II) (3) and Pt(II) (4) yielded bis(phosphine) species [MCl2L2] (5–8). Complexes 5–8 were characterized by 1H, 13C, 31P, 195Pt NMR and HRMS. The structures of all these new complexes were determined with single crystal X-ray diffraction. The configuration of the complexes in a CDCl3 solution was investigated via comparing of 31P NMR data in solution and solid state. | |
| 66 | Y. Kondratenko, T. Kochina, V. Fundamensky, I. Ignatyev, T. Panikorovskii, G. Nyanikova, “Triethanolammonium salicylate – Protic alkanolammonium ionic liquid”, J. Mol. Liquids 2016,221, 1218-1224 DOI:10.1016/j.molliq.2016.06.085 | |
 |
Abstract. Triethanolammonium salicylate was synthesized, characterized by IR and NMR spectroscopy, elemrntal analysis, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). The structure was established, and cation-anion interactions were investigated by X-ray diffraction analysis. It was found that exchange reaction occurs at triethanolammonium salts interaction with silver (I) nitrate, and the product structure was established by X-ray diffraction analysis. The influence of triethanolammonium salicylate solution on the growth properties of the fungus Rhizopus oryzae was studied. | |
| 67 | A. Kulyashova, M. Krasavin, “Convenient modular construction of medicinally important 5-acylamino-4,5-dihydroisoxazoles featuring four elements of diversity”, Tetrahedron Lett. 2016,57, 4395-4397 DOI:10.1016/j.tetlet.2016.08.059 | |
 |
Abstract. An efficient modular approach toward medicinally important 5-acylamino-4,5-isoxazolines via the 1,3-dipolar cycloaddition reaction of nitrile oxides and MCR-derived enamide building blocks is described. This approach results in isoxazolines containing four elements of diversity utilizing two practically simple synthetic operations. | |
| 68 | A.G. Larina, V.E. Nosova, A.S. Filatov, A.P. Molchanov, G.L. Starova, A.A. Zolotarev, V.M. Boitsov, A.V. Stepakov, “The first example of the reactions of cyclopropenes with N-acyliminium cations generated from hydroxylactams”, Tetrahedron 2016, 72, 5064-5073 DOI:10.1016/j.tet.2016.06.052 | |
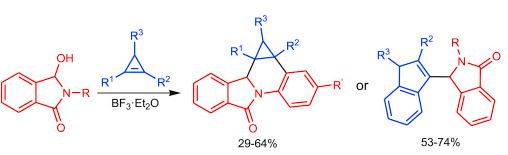 |
Abstract. The first example of the reactions of cyclopropenes with N-acyliminium cations is described. 3-(1H-Inden-3-yl)isoindolin-1-ones and cyclopropa[c]isoindolo[2,1-a]quinolones were prepared by BF3·OEt2 mediated reactions of cyclopropenes with the N-acyliminium cations, generated from 2-aryl(benzyl)-3-hydroxyisoindolin-1-ones. | |
| 69 | Maria S. Ledovskaya, Alexander P. Molchanov, Rafael R. Kostikov, Taras L. Panikorovsky, Vladislav V. Gurzhiy, Mikhail N. Ryazantsev, Vitali M. Boitsov, Alexander V. Stepakov, “Anthracene-fused isoxazolopyrrolo[2,1-a]isoquinolines via an endocyclic N-acyliminium ion cyclization: a joint experimental andtheoretical study”, Tetrahedron 2016, 72, 4827-4834 DOI:10.1016/j.tet.2016.06.048 | |
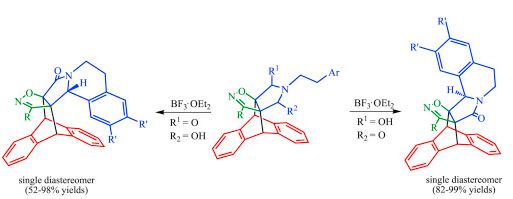 |
Abstract. A simple and efficient strategy is reported for the synthesis of anthracene-fused isoxazolopyrrolo[2,1-a]isoquinolines via an endocyclic N-acyliminium ion cyclization. The cyclization of 18-aryl-21-(2-arylethyl)-22-hydroxy-16-oxa-17,21-diazahexacyclo[6.6.5.315,19.02,7.09,14.015,19]docosa-2,4,6,9,11,13,17-heptaen-20-ones proceeds with high stereoselectivity, leading to 28-aryl-30-oxa-12,29-diazaoctacyclo[13.6.6.32,14.02,14.03,12.04,9.016,21.022,27]triaconta-4,6,8,16,18,20,22,24,26,28-decaen-13-ones. The N-acyliminium cyclization of 18-aryl-21-(2-arylethyl)-20-hydroxy-16-oxa-17,21-diazahexacyclo[6.6.5.315,19.02,7.09,14.015,19]docosa-2,4,6,9,11,13,17-heptaen-22-ones occurs only for substrates with electron-rich aromatic groups in the arylalkyl fragment. In these cases, cyclization also proceeds with a high stereoselectivity with the formation of anthracene-fused isoxazolopyrrolo[2,1-a]isoquinolines as single diastereomers. To understand the mechanisms that allow for cyclization of N-acyliminium ion a quantum chemical investigation was performed. | |
| 70 | A. Lepikhina, O. Bakulina, D. Dar’in, M. Krasavin, “The first solvent-free synthesis of privileged g- and d-lactams via the Castagnoli–Cushman reaction”, RSC Adv. 2016, 6, 83808-83813 DOI:10.1039/c6ra19196g | |
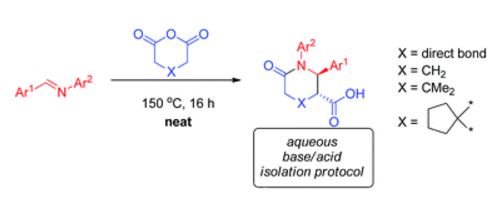 |
Abstract. The first solvent-free protocol for the atom-economical Castagnoli–Cushman reaction is reported. It substantially broadens the reaction scope with respect to achievable substitution patterns around the privileged lactam cores compared to the traditional reaction format employing aromatic hydrocarbon solvents. The convenient product isolation protocol involves the use of aqueous base and acid solutions. | |
| 71 | S.V. Lozovskiy, A.S. Bogachenkov, A.V. Dogadina, A.V. Vasilyev, “Acid-promoted transformations of aryl substituted diphenylphosphoryl allenes”, Tetrahedron Lett. 2016, 57, 3167-3170 DOI:10.1016/j.tetlet.2016.06.026 | |
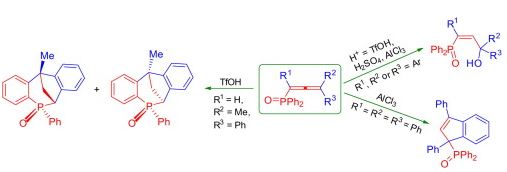 |
Abstract. 1-(Diphenylphosphoryl)alka-1,2-dienes, bearing aryl substituents at the allene system, under the action of Brønsted (TfOH, H2SO4) or Lewis (AlCl3) acids gave rise to 3-hydroxyalk-2-en-1-yl-diphenylphosphine oxides (diphenylphosphoryl allyl alcohols) in yields of 57–98%. In some cases, the formation of (diphenylphosphoryl)indenes and phosphaheterobicyclic structures was observed. | |
| 72 | A.G. Lyapunova, N.A. Danilkina, A.F. Khlebnikov, B. Köberle, S. Bräse, I.A. Balova, “Oxaenediynes through the Nicholas-Type Macrocyclization Approach”, Eur. J. Org. Chem. 2016, 4842-4851 DOI:10.1002/ejoc.201600767 | |
 |
Abstract. The Nicholas-type macrocyclization has been used for the first time for the synthesis of 10- and 11-membered oxaenediynes fused to a benzothiophene. The acyclic starting materials were easily synthesized by electrophilic cyclization of o-(buta-1,3-diynyl)thioanisoles followed by a Sonogashira coupling of the resulting 2-ethynyl-3-iodobenzothiophenes with functionalized alkynes. A high reactivity of the 10-membered oxacycles in the Bergman cyclization was predicted by DFT calculations and confirmed by differential scanning calorimetry. The ability of enediynes to induce single-strand PM2 DNA scissions was also found. | |
| 73 | A.A. Melekhova, A.S. Novikov, N.A. Bokach, M.S. Avdonceva, V.Yu. Kukushkin, “Characterization of Cu-ligand bonds in tris-pyrazolylmethane isocyanide copper(I) complexes based upon combined X-ray diffraction and theoretical study”, Inorg. Chim. Acta 2016, 450, 140-145 DOI:10.1016/j.ica.2016.05.031 | |
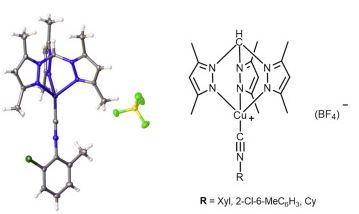 |
Abstract. The complexes [Cu(tpm∗)(CNR)](BF4) (tpm∗ = tris(3,5-dimethylpyrazolyl)methane; R = Xyl [1](BF4), 2-Cl-6-MeC6H3 [2](BF4), Cy [3](BF4)) were prepared by the reaction of [Cu(NCMe)4](BF4) with tpm∗ and CNR (CH2Cl2, 20–25 °C) and these species were characterized by elemental analyses, molar conductivities, high resolution ESI+-MS, IR, 1H and 13C{1H} NMR spectroscopies and also X-ray diffraction for [1](BF4) and [2](BF4). Results of the theoretical DFT (M06/6-31G(d)) study of binding in the copper complexes using NBO and CDA analyses indicate that (i) the influence of crystal packing effects on the geometrical features of [Cu(tpm∗)(CNXyl)]+ cation is insignificant, (ii) the coordination bonds Cu–C in the tris-pyrazolylmethane isocyanide complexes are relatively strong exceeding 50 kcal/mol, and (iii) the CDA data for these systems demonstrate that the {M} ← L σ-donation prevails over the {M} → L π-back-donation. | |
| 74 | V.N. Mikhaylov, V.N. Sorokoumov, K.A. Korvinson, A.S. Novikov, I.A. Balova, “Synthesis and Simple Immobilization of Palladium(II) Acyclic Diaminocarbene Complexes on Polystyrene Support as Efficient Catalysts for Sonogashira and Suzuki−Miyaura Cross-Coupling”, Organometallics 2016, 35, 1684-1697 DOI:10.1021/acs.organomet.6b00144 | |
 |
Abstract. Immobilization of palladium(II) acyclic diaminocarbene (Pd(II)-ADC) complexes on a resin support surface has been easily performed by metal-mediated addition of amino groups of benzhydrylamine-polystyrene to the coordinated isocyanide ligand of cis-PdCl2(CNR)2 (R = t-Bu, Cy). The investigation of the benzhydrylamine reaction with palladium-coordinated isocyanides in solution has revealed that, depending on the reaction conditions, two carbene-type complexes can be obtained as a result of the addition to the CN triple bond, as well as a third complex which is formed via substitution of the isocyanide ligand by benzhydrylamine. Nucleophilic addition of an amino group to the isocyanide ligand has led to a cis-acyclic diaminocarbene complex or a cationic diaminocarbene complex with trans configuration and an intramolecular hydrogen-bonded chloride anion (the nature of this noncovalent interaction was analyzed by DFT calculations, including AIM analysis). The unsupported and resin-supported palladium catalysts have demonstrated high catalytic activity in both Sonogashira and Suzuki–Miyaura cross-coupling. The supported catalyst can be recovered and repeatedly reused without a significant loss in efficiency. The degree of the palladium binding with polystyrene, the oxidation state, and the palladium leaching level were investigated by XPS and XRF analyses. | |
| 75 | A.P. Molchanov, T.Q. Tran, A.V. Stepakov, R.R. Kostikov, “Reductive Cleavage and Subsequent Transformations of 5-Oxa-6-azaspiro[2.4]heptane-1-carboxylates”, Russ. J. Org. Chem. 2016, 52, 404-408 DOI:10.1134/S1070428016030180 | |
| Abstract. Treatment of methyl 4,6-diaryl-5-oxa-6-azaspiro[2.4]heptane-1-carboxylates with zinc in acetic acid leads to cleavage of the N–O bond in the isoxazolidine ring with formation of 1,3-amino alcohols whose subsequent cyclization under the reaction conditions yields bi- or tricyclic lactams or lactones with retention of the three-membered ring. | ||
| 76 | V.A. Nikolaev, J.J. Medvedev, O.S. Galkina, K.V. Azarova, C. Schneider, “Unusual reactions of diazocarbonyl compounds with α,β-unsaturated δ-amino esters: Rh(II)-catalyzed Wolff rearrangement and oxidative cleavage of N–H-insertion products”, Beilstein J. Org. Chem. 2016, 12, 1904-1910 DOI:10.3762/bjoc.12.180 | |
| Abstract. Rh(II)-сatalyzed reactions of aroyldiazomethanes, diazoketoesters and diazodiketones with α,β-unsaturated δ-aminoesters, in contrast to reactions of diazomalonates and other diazoesters, give rise to the Wolff rearrangement and/or oxidative cleavage of the initially formed N–H-insertion products. These oxidation processes are mediated by Rh(II) catalysts possessing perfluorinated ligands. The formation of pyrrolidine structures, characteristic for catalytic reactions of diazoesters, was not observed in these processes at all. | ||
| 77 | L.L. Rodina, O.S. Galkina, G. Maas, M.S. Platz, V.A. Nikolaev, “A New Method for C-H Functionalization of Aliphatic Compounds by an Unusual Photochemical Reaction of Diazoketones without Elimination of Nitrogen”, Asian J. Org. Chem. 2016, 5, 691-698 DOI:10.1002/ajoc.201600050 | |
 |
Abstract. Benzophenone-sensitized reactions of 4-diazotetrahydrofuran-3(2H)-ones with H-donors such as tetrahydrofuran, 1,4-dioxane, cyclohexane and diethyl ether, occur without elimination of nitrogen and give rise to the corresponding N-substituted hydrazones or bis-hydrazonoethanes due to a formal insertion of the terminal N atom of diazo group into α-C−H bonds of ethers and aliphatic hydrocarbons, with yields of up to 78 %. Long-wavelength UV irradiation (λ>310 nm) is most suitable for this process, whereas oxygen molecules adequately quench the triplet excited state of the diazoketone and reduce the preparative yields of C−H-insertion products. Hence this photochemical reaction of diazoketones can be used for C−H functionalization of different aliphatic compounds. | |
| 78 | D.S. Ryabukhin, D.N. Zakusilo, M.O. Kompanets, A.A. Tarakanov, I.A. Boyarskaya, T.O. Artamonova, M.A. Khohodorkovskiy, I.O. Opeida, A.V. Vasilyev, “Superelectrophilic activation of 5-hydroxymethylfurfural and 2,5-diformylfuran: organic synthesis based on biomass-derived products”, Beilstein J. Org. Chem. 2016, 12, 2125-2135 DOI:10.3762/bjoc.12.202 | |
| Abstract. The reaction of 5-hydroxymethylfurfural (5-HMF) with arenes in superacidic trifluoromethanesulfonic acid (triflic acid, TfOH) as the solvent at room temperature for 1–24 h gives rise to 5-arylmethylfurfurals (yields of 17–91%) and 2-arylmethyl-5-(diarylmethyl)furans (yields of 10–37%). The formation of these two types of reaction products depends on the nucleophilicity of the arene. The same reactions under the action of acidic zeolites H-USY in high pressure tubes at 130 °C for 1 h result in the formation of only 5-arylmethylfurfurals (yields of 45–79%). 2,5-Diformylfuran (2,5-DFF) in the reaction with arenes under the action of AlBr3 at room temperature for 1 h leads to 5-(diarylmethyl)furfurals (yields of 51–90%). The reactive protonated species of 5-HMF and 2,5-DFF were characterized by NMR spectroscopy in TfOH and studied by DFT calculations. These reactions show possibilities of organic synthesis based on biomass-derived 5-HMF and 2,5-DFF. | ||
| 79 | A. Sapegin, V. Panova, E. Reutskaya, A.V. Smirnov, M. Krasavin, “A novel, flexible strategy to construct privileged dibenzo[b,f][1,4,5] oxathiazepine 5,5-dioxides and their heterocyclic isosteres”, Tetrahedron 2016, 72, 7570-7578 DOI:10.1016/j.tet.2016.10.008 | |
 |
Abstract. Secondary o-hydroxybenzene sulfonamides have been explored as bis-electrophilic partners in a practically simple, atom-economical approach to dibenzo[b,f][1,4,5]oxathiazepine-5,5-dioxides and their heterocyclic analogs, which is an underexplored version of privileged tricyclic scaffolds for drug design. The reaction proceeds smoothly and regiospecifically under conventional heating conditions and delivers the target compounds in good to excellent yields. The approach represents a completely new disconnection of the dibenzo[b,f][1,4,5]oxathiazepine-5,5-dioxide scaffolds and allows accessing a new chemical space in terms of the substitution pattern. In particular, heterocycles can be conveniently varied within the tricyclic frameworks by altering the nature of the bis-electrophilic aromatic partner in the cyclization. This has been demonstrated by the synthesis of three hitherto undescribed heterotricyclic scaffolds. The cyclizations were shown to proceed via the Smiles rearrangement, as had been observed by us for a range of similar ring-forming processes. | |
| 80 | E.T. Satumov, J.J. Medvedev, D.I. Nilov, M.A. Sandzhieva, I.A. Boyarskaya, V.A. Nikolaev, A.V. Vasilyev, “Protonation and transformations of a-diazo-b-dicarbonyl compounds in superacids: generation of the strongest carbon-centered cationic electrophiles at the protonation of diazomalonates in FriedeleCrafts reactions”, Tetrahedron 2016, 72, 4835-4844 DOI:10.1016/j.tet.2016.06.051 | |
 |
Abstract. Protonation of diazodiketones N2C(COR)2 in Brønsted superacids (TfOH, FSO3H, TfOH–SbF5) gives rise to stable and non-reactive O,O-diprotonated at carbonyl oxygens species N2C(C(double bond; length as m-dashOH+)R)2, which were studied by means of 1H and 13C NMR. Diazomalonates N2C(CO2Alk)2, contrary to diazodiketones, react with TfOH or HF, releasing nitrogen and producing triflates of oxymalonates TfOCH(CO2Alk)2 or fluoromalonates FCH(CO2Alk)2, respectively. Diazoketoesters N2C(COR)(CO2Alk) react in the same way only with TfOH, but not with HF. The reactions of diazomalonates with arenes ArH (benzene, toluene, xylenes) in TfOH solution yield corresponding Friedel–Crafts reaction products ArCH(CO2Alk)2. According to performed DFT calculations, trication +CH(C(double bond; length as m-dashOH+)OMe)2, a possible intermediate, which is derived from protonation of dimethyl diazomalonate, should be the strongest cationic carbon-centered electrophile known up to date. | |
| 81 | S. Saulnier, A.A. Golovanov, A.V. Vasilyev, “A controlled tandem transformation of conjugated enynones with arenes under superelectrophilic activation leading to aryl substituted dienones and indenes”, RSC Adv. 2016, 6, 103546-103555 DOI:10.1039/c6ra21965a | |
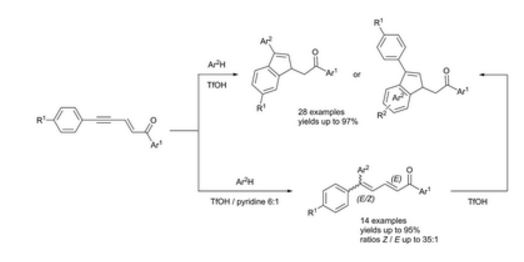 |
Abstract. Under superelectrophilic activation in triflic acid (TfOH) and in the presence of arenes, 1,5-diarylpent-2-en-4-yn-1-ones lead to aryl-substituted indenes by addition of arene to the acetylenic bond followed by an intramolecular cyclization in a tandem process, with yields up to 97%. The regiochemistry of product formation is predicted and controlled by varying the aromatic substituents on the starting materials, which makes it possible to obtain diversely substituted indenes. The use of pyridine as a co-solvent of the superacid TfOH allows control of the extent of the tandem transformation and to isolate the aryl-conjugated dienone intermediates before cyclization with high E/Z diastereoselectivities and yields up to 95%. The reaction mechanism is highlighted and involves cationic species that have been observed and characterized by NMR. | |
| 82 | A.Y. Timoshkin, A.S. Lisovenko, D.A. Doinikov, I.V. Kazakov, A.S. Zavgorodnii, “Donor–acceptor complexes of inorganic analogs of benzene”, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 591-596 DOI:10.1080/10426507.2015.1128913 | |
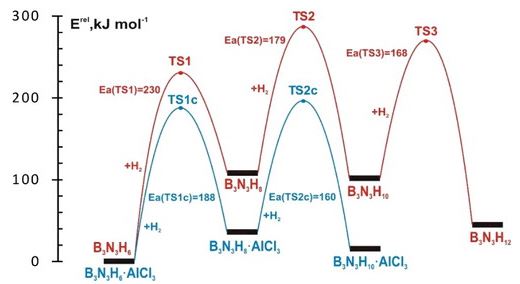 |
Abstract. Results of experimental and theoretical studies of inorganic benzene analogs: borazine, substituted borazines, polyborazines, alumazene, and their donor–acceptor complexes are summarized. Structural and energetic aspects of complex formation and thermal stability of heterocycles and their complexes are discussed. A mechanism for the gas-phase acetonitrile polymerization in the presence of alumazene is proposed on the basis of computational studies. It is experimentally shown that solution of B,B′,B″-tribromborazine in deuterobenzene undergoes fast (within minutes) H/D exchange in the presence of Lewis acid AlBr3. The proposed electrophilic substitution mechanism for the exchange is supported by quantum-chemical computations. In contrast, in the presence of AlBr3, unsubstituted borazine in deuterobenzene polymerizes with hydrogen evolution without H/D exchange. The absence of the H/D exchange may be explained by larger stability of the borazonium ion B3N3H7+ which prevents operation of the catalytic cycle. Quantum-chemical computations at B3LYP/TZVP level of theory indicate that upon complexation with AlCl3 both endothermicity and activation energies of hydrogenation processes of borazine and polyborazines are significantly reduced. The use of Lewis acids as catalysts in the processes of regeneration of spent hydrogen fuel is recommended. | |
| 83 | A. Vereshchagin, V. Sizov, M. Verjuzhskij, S. Hrom, A. Volkov, J.S. Danilova, M. Novozhilova, A. Laaksonen, O.V. Levin, “Interaction of amines with electrodes modified by polymeric complexes of Ni with salen-type ligands”, Electrochimica Acta, 2016, 211, 726-734 DOI:10.1016/j.electacta.2016.06.087 | |
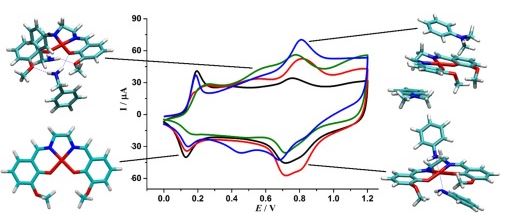 |
Abstract.Chemically modified electrodes based on complexes of transition metals with N,N′-ethylenebis(salicylimine) (salen) type ligands are reported to act as redox mediators due to axial coordination of exogeneous ligands to metal atom in oxidized form of complex. Thus such electrodes can be used as amperometric and voltammetric detectors of various compounds. However, effects of ligand structure and ligand-analyte specific interactions on the electrochemical response of said complexes have not been studied yet for organic analytes. In this article we demonstrate that some aromatic amines can interact with salen-type ligands by several routes, depending on the ligand and amine structure. It leads to the ligand polarization, and thus to the unique shape of voltammetric curves of chemically modified electrodes, based on polymeric salen complexes, immersed in amines solutions. Such feature allows using the electrodes as sensors for molecular recognition. The same electrodes can act as electron transfer catalysts of the amines oxidation, and may be applicable to the quantitative analysis of amines with the detection limit about 10 μmol l−1. | |
| 84 | M.S. Ledovskaya, A.P. Molchanov, R.R. Kostikov, T.L. Panikorovsky, V.V. Gurzhiy, M.N. Ryazantsev, V.M. Boitsov, AV. Stepakov, “Anthracene-fused isoxazolopyrrolo[2,1-a]isoquinolines via an endocyclic N-acyliminium ion cyclization: a joint experimental and theoretical study”, Tetrahedron 2016, 72, 4827-4834 DOI:10.1016/j.tet.2016.06.048 | |
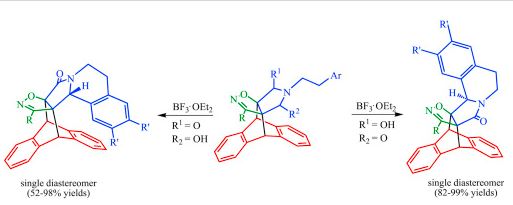 |
Abstract.A simple and efficient strategy is reported for the synthesis of anthracene-fused isoxazolopyrrolo[2,1-a]isoquinolines via an endocyclic N-acyliminium ion cyclization. The cyclization of 18-aryl-21-(2-arylethyl)-22-hydroxy-16-oxa-17,21-diazahexacyclo[6.6.5.315,19.02,7.09,14.015,19]docosa-2,4,6,9,11,13,17-heptaen-20-ones proceeds with high stereoselectivity, leading to 28-aryl-30-oxa-12,29-diazaoctacyclo[13.6.6.32,14.02,14.03,12.04,9.016,21.022,27]triaconta-4,6,8,16,18,20,22,24,26,28-decaen-13-ones. The N-acyliminium cyclization of 18-aryl-21-(2-arylethyl)-20-hydroxy-16-oxa-17,21-diazahexacyclo[6.6.5.315,19.02,7.09,14.015,19]docosa-2,4,6,9,11,13,17-heptaen-22-ones occurs only for substrates with electron-rich aromatic groups in the arylalkyl fragment. In these cases, cyclization also proceeds with a high stereoselectivity with the formation of anthracene-fused isoxazolopyrrolo[2,1-a]isoquinolines as single diastereomers. To understand the mechanisms that allow for cyclization of N-acyliminium ion a quantum chemical investigation was performed. | |
| 85 | A.V. Hubina, A.A. Pogodaev, V.V. Sharoyko, E.G. Vlakh, T.B. Tennikova, “Self-assembled spin-labeled nanoparticles based on poly(amino acids)”, Reactive and Functional Polymers 2016, 100, 173–180 DOI:10.1016/j.reactfunctpolym.2016.01.018 | |
 |
Abstract.The development of detectable nanoparticles for controlled drug delivery systems has tremendous practical importance regarding the monitoring of drug pathway in organism. Self-assembly amphiphilic block-copolymer poly(l-glutamic acid)-b-poly(l-phenylalanine) (pGlu-b-pPhe) was chosen for the preparation of discussed nanoparticles. The synthesis of blocks was carried out using ring-opening polymerization (ROP) of N-carboxyanhydrides of mentioned amino acids. To introduce the spin label at C-terminal position of hydrophilic block, (4-amino-2,2,6,6-tetramethylpiperidin-1-yl)oxyl (4-amino-TEMPO) was applied as ROP initiator and the polymerization of hydrophobic block was carried out with previously synthesized macroinitiator. The results obtained by transmission electron microscopy clearly showed that TEMPO-pGlu-b-pPhe polymer was really capable to self-assembling in aqueous solutions followed by polymersome formation. The mean size of nanoparticles was increased in a range of TEMPO-pGlu43-b-pPhe12 < TEMPO-pGlu43-b-pPhe29 < TEMPO-pGlu43-b-pPhe49 as 60 < 200 < 280 nm, respectively. EPR spectroscopy of the solutions of spin-labeled homopolymer TEMPO-p-γ-Glu(Bzl), block copolymers TEMPO-p-γ-Glu(Bzl)-b-pPhe and suspension of polymersomes formed from TEMPO-p-Glu-b-pPhe was performed and the results were compared. It was proved that in the case of nanoparticles EPR detectable spin labels are located on polymersome surface. The experiments in cell culture demonstrated the absence of cytotoxicity of labeled nanoparticles. Additionally, it was shown that TEMPO-label can be detected inside the cell by EPR method. | |
| 86 | M. V. Il’in, D. S. Bolotin, M. Ya. Demakova, and M. S. Avdontseva, “Structure of Aminonitrones and Electronic Effect of Substituents on Their Acid-Base Properties”, Russian Journal of General Chemistry, 2016, 86, 806–809 DOI:10.1134/S1070363216040071 | |
| Abstract.A series of para-substituted aromatic aminonitrones p-RC6H4C(NH2)=N+(Me)O– (R = NMe2, H, Br, Cl, CF3) have been prepared. Acidity constants of the conjugate acids RC6H4C(NH2)N+(Me)OH at 25°C in a EtOH–H2O mixture (5: 95) have been determined by potentiometric titration. A linear correlation between log (kR/kH) and σpara values has been revealed, and a ρ298(σpara) parameter has been determined as of 0.635. | ||
| 87 | Mikhail A. Kinzhalov, Svetlana A. Timofeeva, Konstantin V. Luzyanin, Vadim P. Boyarskiy, Anton A. Yakimanskiy, Matti Haukka, and Vadim Yu. Kukushkin, “Palladium(II)-Mediated Addition of Benzenediamines to Isocyanides: Generation of Three Types of Diaminocarbene Ligands Depending on the Isomeric Structure of the Nucleophile”,Organometallics 2016, 35, 218−228 DOI:10.1021/acs.organomet.5b00936 | |
 |
Abstract.Coupling of the palladium-bis(isocyanide) complexes cis-[PdCl2(CNR)2] (R = 2,6-Me2C6H3 1, 2-Cl-6-MeC6H3 2) with benzene-1,3-diamine (BDA1) leads to the diaminocarbene species cis-[PdCl2(CNR){C(NHR)═NH(1,3-C6H4NH2)}] (5 and 6, respectively). In this reaction, BDA1 behaves as a monofunctional nucleophile that adds to one of the RNC ligands by one amino group. By contrast, the reaction of 1 and 2 with benzene-1,4-diamine (BDA2) involves both amino functionalities of the diamine and leads to the binuclear species [cis-PdCl2(CNR){μ-C(NHR)═NH(1,4-C6H4)NH═C(NHR)}-(cis)-PdCl2(CNR)] (6 and 7) featuring two 1,4-bifunctional diaminocarbene ligands. The reaction of cis-[PdCl2(CNR)2] (R = cyclohexyl 3) with either BDA1 or BDA2 does not afford any isolable carbene derivatives. The most versatile chemistry was observed when 1–3 were treated with benzene-1,2-diamine (BDA3) and the relevant substituted 1,2-diamines, viz., 4,5-dimethylbenzene-1,2-diamine (BDA4) and 4,5-dichlorobenzene-1,2-diamine (BDA5). The addition of these diamines brings about the formation of the monocarbene cationic complexes cis-[PdCl(CNR){C(NHR)═NHC6H2X2NH2}]Cl (X = H, Me, Cl) (8–16), the Chugaev-type C,C-bound bis-carbenes cis-[PdCl2{C(NHR)═NHC6H2X2NH═C(NHR)}] (17, 18), and the bis(C,N-chelated)carbene complexes cis-[Pd{C(NHR)═NHC6H2X2NH2}2]Cl2 (19–24). All prepared complexes (with the exception of 17 and 18) were isolated as colorless or pale yellow solids and characterized by elemental analyses (C, H, N), HRESI±-MS, IR, 1H and 13C{1H} NMR spectroscopies, and 4, 7, 13, 16, and 24 by X-ray diffraction. Complexes 17 and 18 were characterized by HRESI±-MS and IR spectroscopy, and their structures were established by X-ray crystallography. | |
| 88 | A. A. Petrov, V. V. Pakal’nis, I. V. Zerova, and S. I. Yakimovich, “Reactions of 2-Perfluoroacylcycloalkanones with Benzoylhydrazine”, Russian Journal of Organic Chemistry, 2016, Vol. 52, No. 2, pp. 285–288 DOI:10.1134/S1070428016020202 | |
| 89 | A. V. Zerov, T. S. Krupenya, A. A. Petrov, and S. I. Yakimovich, “Reaction of Trifluoromethyl-Containing 1,3-Dicarbonyl Compounds with Bis-hydrazides”, Russian Journal of Organic Chemistry, 2016, 52, 312–318 DOI:10.1134/S1070428016030039 | |
| Abstract.Carbonic dihydrazide reacted with 2 equiv of trifluoroacetylacetone to give a compound containing 5-hydroxy-4,5-dihydropyrazole and keto enehydrazine fragments. Analogous 2 : 1 condensations of trifluoro-acetylacetone and 4-ethoxy-1,1,1-trifluorobut-3-en-2-one with oxalohydrazide and malonohydrazide afforded bis(5-hydroxy-4,5-dihydropyrazole) structures as two diastereoisomers. | ||
| 90 | T.L. Panikorovskii, S.V. Krivovichev, E.V. Galuskin, V.V. Shilovskikh, A.S. Mazur, A.V. Bazai, “Si-deficient, OH-substituted, boron-bearing vesuvianite from Sakha-Yakutia, Russia: a combined single-crystal, 1H MAS-NMR and IR spectroscopic study”, Eur. J. Mineral., 2016, 28, 931–941 DOI:10.1127/ejm/2016/0028-2570 | |
 |
Abstract.Single crystals of Si-deficient vesuvianite with significant degree of hydrogarnet-type (SiO4)4−–(O4 H4)4− substitution occur as epitactic overgrowth on the surface of large wiluite crystals from the Wiluy River, Sakha–Yakutia, Russia. Electron-microprobe analysis revealed considerable Si-deficiency, Si ranging from 16.30 to 17.50 apfu. The crystal structure of the mineral has been refined in the P4/nnc space group, a = 15.5876(4), b = 11.8021(5) Å to R 1 = 0.028 for 1533 unique observed reflections. The refinement of the site-occupancy factors confirmed significant vacancy at the Z(1) and Z(2) sites (27% and 10%, respectively), with associated increase of the Z(1)–O and Z(2)–O bond lengths to 1.697 and 1.655 Å, respectively. The increased size of the Z(1) tetrahedra results in the compression of the X(1) polyhedra, while the expansion of the Z(2) tetrahedra is compensated by the compression of the X(4) polyhedra. As a result, the significant degree of the hydrogarnet-type substitutions does not have an essential influence upon the unit-cell parameters compared to the usual defect-free vesuvianite. Thermogravimetric (TGA) and differential scanning calorimetry (DSC) curves indicated two steps of the weight loss in the temperature ranges of 600–900°C (2.08 wt%) and 1014°C (2.22 wt%). The total weight loss is estimated as 4.30%, which is in good agreement with the total content of OH estimated as 13.72 apfu. The 1H solid state MAS-NMR demonstrates the presence of a strong additional line at 3.38 ppm compared to the usual vesuvianite, which is explained by the existence of the additional H(3) site. The infrared spectrum in the OH-stretching vibration region shows the additional (B′) absorption band at 3618 cm−1 typical for a hydrogarnet-type substitution. | |
| 91 | V.V. Tomaev, A.S. Mazur, A.S. Grevtsev, “A study of the process of thermal oxidation of lead selenide by the NMR and XRD methods”, Glass Phys. Chem., 2017, 43, 70–74 DOI:10.1134/S1087659617010163 | |
 |
Abstract.Studies of the process of oxidation of powder samples of lead selenide in a dry air atmosphere have been performed. As shown by the methods of X-ray diffraction and scanning electron microscopy, the process of thermal treatment of samples resulted in the formation of the PbSeO3 phase, aside from the initial PbSe phase. Studies by the method of nuclear magnetic response (NMR) allowed revealing the dynamics of changes in the spectrum under the changed thermal treatment conditions. | |