2017 |2016 | 2015 | 2014 | 2013 | 2013-2012
| № | Abstract | |
| 1 | M.Ya. Demakova, D.S. Bolotin, N.A. Bokach, G.L. Starova, V.Yu. Kukushkin, “Metal-mediated cyanamide–hydroxyguanidine coupling”, Inorg Chim. Acta, 2015, 425, 114-117 DOI:10.1016/j.ica.2014.10.015 | |
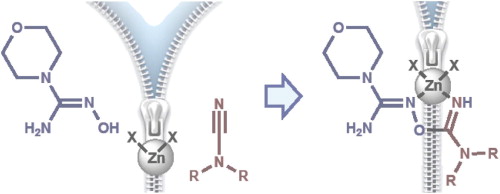 |
Abstract. Reaction of the cyanamides R2NCN (R = Me 2a, Et 2b) with the hydroxyguanidine OC4H8NC(=NOH)NH2 (1) in the presence of zinc halides leads to [ZnX2{HN=C(NR2)ON=C(NH2)NC4H8O}] derived from the ZnII-mediated cyanamide–hydroxyguanidine coupling. This reaction is the first observation of interplay between any nitrile group and any hydroxyguanidine both in metal-involving and metal-free chemistry. Complexes 3a,b–5a,b rather rapidly degrade in solutions at RT, but solvates 3a,b·MeOH and 4a·MeOH are sufficiently stable in the solid state and they were characterized by IR, HRESI+-MS, solid state CP-MAS TOSS 13C NMR, and X-ray crystallography (for 3a,b). Complexes 4b and 5a,b were identified in solutions by HRESI+-MS. | |
| 2 | A. Ya. Bespalov, T. L. Gorchakova, A. Yu. Ivanov, M. A. Kuznetsov, L. M. Kuznetsova, A. S. Pankova, L. I. Prokopenko, M. S. Avdontceva, «Alkylation and Aminomethylation of 1,3-Dihydro-2Н-Benzimidazole-2-Thione», Chem. Heterocycl. Compd., 2015, 50(11), ASAP DOI:10.1007/s10593-014-1623-z | |
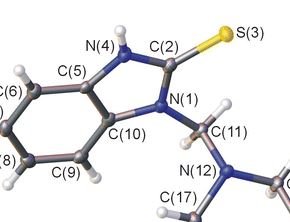 |
Abstract. Alkylation of 1,3-dihydro-2Н-benzimidazole-2-thione (2-mercaptobenzimidazole) with bromoethane and chloroacetic acid derivatives occurrs at the sulfur atom, leading to the corresponding 2-sulfanylbenz-imidazole derivatives. Aminomethylation of 1,3-dihydro-2Н-benzimidazole-2-thione with piperidine and 4-methylpiperidine gives reaction products at both nitrogen atoms, while reaction with morpholine gives derivative at only one nitrogen atom, which is in an equilibrium with the starting compound and bis-adduct in DMSO solution. | |
| 3 | S. Pylaeva, C. Allolio, B. Koeppe, G.S. Denisov, H.-H. Limbach, D. Sebastiani, P.M. Tolstoy, “Proton transfer in hydrogen bonded complex caused by solvation shells fluctuations: ab initio MD study of anionic phenolate-carboxylic acid and neutral pyridine-carboxylic acid systems”, Phys. Chem. Chem. Phys., 2015, 17, 4634-4644 DOI:10.1039/c4cp04727c | |
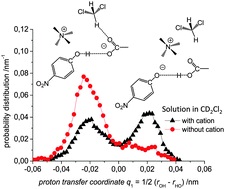 |
Abstract. We present a joint experimental and quantum chemical study on the influence of solvent dynamics on the protonation equilibrium in a strongly hydrogen bonded phenol–acetate complex in CD2Cl2. Particular attention is given to the correlation of the proton position distribution with the internal conformation of the complex itself and with fluctuations of the aprotic solvent. Specifically, we have focused on a complex formed by 4-nitrophenol and tetraalkylammonium–acetate in CD2Cl2. Experimentally we have used combined low-temperature 1H and 13C NMR and UV-vis spectroscopy and showed that a very strong OHO hydrogen bond is formed with proton tautomerism (PhOH⋯−OAc and PhO−⋯HOAc forms, both strongly hydrogen bonded). Computationally, we have employed ab initio molecular dynamics (70 and 71 solvent molecules, with and without the presence of a counter-cation, respectively). We demonstrate that the relative motion of the counter-cation and the “free” carbonyl group of the acid plays the major role in the OHO bond geometry and causes proton “jumps”, i.e. interconversion of PhOH⋯−OAc and PhO−⋯HOAc tautomers. Weak H-bonds between CH(CD) groups of the solvent and the oxygen atom of carbonyl stabilize the PhOH⋯−OAc type of structures. Breaking of CH⋯O bonds shifts the equilibrium towards PhO−⋯HOAc form. | |
| 4 | E.Yu. Tupikina, G.S. Denisov, P.M. Tolstoy, “NMR Study of CHN Hydrogen Bond and Proton Transfer in 1,1-dinitroethane Complex with 2,4,6-trimethylpyridine “, J. Phys. Chem. A, 2015, 119, 659-668 DOI:10.1021/jp511493m | |
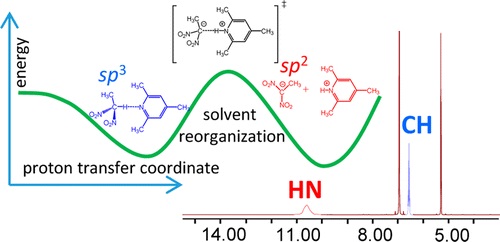 |
Abstract. The intermolecular complex with a CHN hydrogen bond formed by 1,1-dinitroethane (DNE) and 2,4,6-trimethylpyridine (collidine) dissolved in CD2Cl2 was studied experimentally by 1H NMR spectroscopy at 180–300 K. Equilibrium between the molecular CH···N form and the zwitterionic C–/HN+ form was detected in the slow exchange regime in the NMR time scale. No sign of a direct C–···HN+ bond was observed; the ion pair is likely to be held by Coulomb interactions. Moreover, there are indications that the protonated base is involved in the formation of homoconjugated (NHN)+ collidine–collidinium hydrogen bonded complexes. The reaction pathway of proton transfer in the DNE–pyiridine complex in a vacuum was studied computationally at the B3LYP/6-31++G(d,p) level of theory. NMR chemical shifts and coupling constants were calculated for a series of snapshots along the proton transfer coordinate. While the central carbon atom has a pyramidal (sp3) configuration in DNE, it is flat (sp2) in the DNE carbanion. As a result, the most indicative computed NMR parameter reflecting hybridization of a carbon atom appeared to be 1JCC, which starts to change rapidly as soon as a structure with a quasi-symmetric C··H··N bond is reached. Couplings within the hydrogen bridge, 1JCH, 1hJHN, and 2JCN, can serve as good indicators of the degree of proton transfer. | |
| 5 | M.S. Novikov, A.F. Khlebnikov, N.V. Rostovskii, S. Tcyrulnikov, A.A. Suhanova, K.V. Zavyalov, D.S. Yufit, “ Pseudopericyclic 1,5- versus Pericyclic 1,4- and 1,6-Electrocyclization in Electron-Poor 4‑Aryl-2-azabuta-1,3-dienes: Indole Synthesis from 2H‑Azirines and Diazo Compounds”, J Org. Chem. 2015, 80, 18-29 DOI:10.1021/jo501051n | |
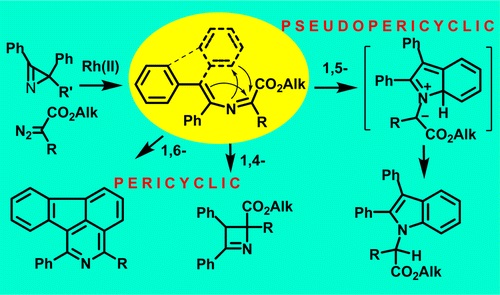 |
Abstract. Transformations of 2-azabuta-1,3-dienes, formed in Rh2(OAc)4-catalyzed reactions of diazo carbonyl compounds with 2H-azirines, dramatically depend on the nature of substituents. 4,4-Diphenyl-2-azabuta-1,3-dienes with two electron-acceptor substituents at C1 undergo thermal 1,5-cyclization to give indoles in good yields. The increase in electron-withdrawing ability of C1-substituents facilitates the reaction that proceeds via pseudopericyclic 1,5-electrocyclization of 2-azabutadiene into 7aH-indolium ylide followed by prototropic shift. 3,4-Diphenyl-2-azabuta-1,3-dienes, resulting from reaction of 2,3-diphenyl-2H-azirine and diazo compounds, do not produce indoles via 1,5-cyclization due to the trans-configuration of the 4-Ph-group and the nitrogen, but undergo 1,4-cyclization to 2,3-dihydroazetes. 1,6-Cyclization into 2H-1,4-oxazines with participation of the oxygen of ester or amide group at C1 of corresponding 2-azabuta-1,3-dienes does not take place due to kinetic and thermodynamic reasons. Instead of this, 1,6-electrocyclization with participation of phenyl substituent at C4 of the 2-azabuta-1,3-dienes, providing isoquinoline derivatives, can occur at elevated temperatures. The DFT-calculations (mPWB1K/6-31+G(d,p)) confirm the dependence of 2-azabuta-1,3-diene transformation type on the nature of substituents. | |
| 6 | A. Penkova, G. Polotskaya, A. Toikka, “Pervaporation composite membranes for ethyl acetate production”, Chem. Eng. Process., 2015, 87, 81-87 DOI:10.1016/j.cep.2014.11.015 | |
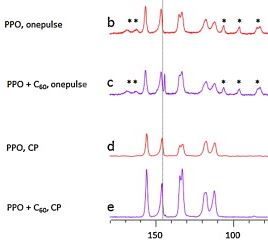 |
Abstract. This paper continues research on the physicochemical features of nanocomposites of poly(2,6-dimethyl-1,4-phenylene oxide) (PPO) and fullerene C60 with the purpose of using them as a selective layer of composite membranes in pervaporation coupling esterification of acetic acid with ethanol to produce ethyl acetate. The C60–PPO/MFFC composite membranes containing up to 3 wt%C60 were prepared in this work. SEM was employed in visualizing the internal morphology of the membranes. The nature of the interaction between PPO and C60 molecules in composite was studied by NMR. TG analysis reveals the fact of increasing thermal destruction point of the membrane due to certain structural changes of the PPO matrix upon incorporation of the C60 molecules. Transport properties of composite membranes with a selective layer containing up to 3 wt%C60 were studied in the pervaporation of the quaternary mixture: ethanol, acetic acid, ethyl acetate, and water in order to identify the availability of their use in the hybrid process “esterification + pervaporation”. The strong permeability enhancement due to the introduction of C60 particles in the PPO network was observed whereas the selectivity was almost steady. The best complex of transport properties was obtained from the 2%C60–PPO/MFFC membrane. | |
| 7 | A.I. Solomatina, D.V. Krupenya, V.V. Gurzhiy, I. Zlatkin, A.P. Pushkarev, M.N. Bochkarev, N.A. Besley, E. Bichoutskaia, S.P. Tunik, “Cyclometallated platinum(II) complexes containing NHC ligands; synthesis, characterization, photophysics and their application as emitters in OLEDs”, Dalton Trans. 2015, 44, 7071-7688 DOI:10.1039/c4dt03106g | |
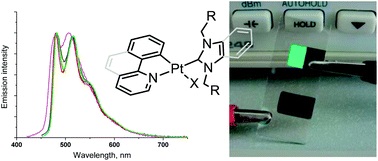 |
Abstract. A series of square planar [Pt(N^C)(NHC)L] complexes containing cyclometallated N^C ligands (phenylpyridine and benzoquinoline) and N-heterocyclic carbene (NHC) – N^C = 2-phenylpyridine, 7,8-benzoquinoline; NHC = 1,3-dibenzylbenzimidazolium, 1,3-diethylbenzimidazolium, 1,3-dibenzylimidazolium; L = Cl, Br, –C2Ph – have been synthesized in moderate to good yields. The complexes obtained were characterized using chemical analysis, MS-ESI spectrometry, NMR spectroscopy and X-ray crystallography. The complexes display moderate to strong phosphorescence in solution (Q.Y. 0.3–7.9%) and in the solid state (Q.Y. 2.7–16.0%), which is related to metal modulated intraligand π–π* transitions located at the aromatic system of cyclometallated ligands with some contribution of the MLCT excited state. Emission lifetimes fall in the range of 0.2–1.5 μs in solution and amount up to 13 μs in the solid state. Analysis of the spectroscopic data together with the density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations clearly support this assignment and show negligible contribution of the auxiliary ligands to the emissive excited states. The compounds obtained were also used to prepare organic light emitting diode (OLED) devices, which display good luminance efficiency emitting in the green area of the visible spectrum. | |
| 8 | A.K. Sánchez Lafarga, F.P. Pacheco Moisés, A. Gurinov, G.G. Ortiz, G.G. Carbajal Arízaga, “Dual responsive dysprosium-doped hydroxyapatite particles and toxicity reduction after functionalization with folic and glucuronic acids”, Mat. Sci. Eng. C, 2015, 48, 541-547 DOI:10.1016/j.msec.2014.12.033 | |
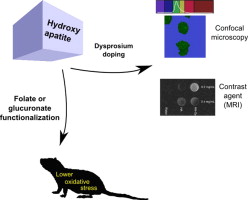 |
Abstract. The development of probes for biomedical applications demands materials with low toxicity levels besides fluorescence or magnetic properties to be detected by confocal microscopes or MRI resonators. Several drug delivery systems or other biomedical materials prepared with hydroxyapatite have been proposed, however, toxicity effects might arise when the size of particles is nanometric. In this study, hydroxyapatite functionalized with glucuronic or folic acids presented lower oxidative stress, measured from lipoperoxides and nitric oxide indicators in rats than pure hydroxyapatite. In separated experiments, hydroxyapatite was doped with dysprosium cations by coprecipitation producing a single crystal phase with fluorescent properties easily visualized by confocal microscopy when excited at 488 nm. These particles also presented the ability to modify the proton relaxation time in T1 maps collected by magnetic resonance imaging. These modified hydroxyapatite nanoparticles could be candidates to design bimodal probes with low toxicity. | |
| 9 | A.S. Bogachenkov, A.V. Dogadina, V.P. Boyarskiy, A.V. Vasilyev, “Acid-promoted transformations of 1-(diphenylphosphoryl)allenes: synthesis of novel 1,4-dihydrophosphinoline 1-oxides”, Org. Biomol. Chem. 2015, 13, 1333-1338 DOI:10.1039/c4ob02269f | |
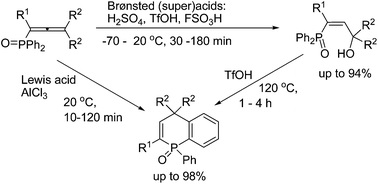 |
Abstract. 1-(Diphenylphosphoryl)alka-1,2-dienes (phosphonoallenes) in Brønsted (super)acids (TfOH, FSO3H, and H2SO4) at −70 to 120 °C for 30 min to 4 h gave, at first, (3-hydroxyalk-1-en-1-yl)diphenylphosphine oxides, as kinetically favorable reaction products, that are further converted into 1-phenyl-1,4-dihydrophosphinoline 1-oxides as thermodynamically stable compounds. The latter compounds are formed from phosphonoallenes under the action of a strong Lewis acid AlCl3 at room temperature for 10–120 min. This is a novel, simple and efficient (short reaction time, high yields) method for synthesis of such 1,4-dihydrophosphinoline 1-oxides. | |
| 10 | D. Boyarskaya, M. Avdontceva, T. Chulkova, “Synthesis and crystal structure of 2-isocyano-4-methylphenyl diphenylacetate: a rare case of an easily accessible odourless isocyanide”, Acta Cryst. C 2015, C71, 155-158 DOI:10.1107/S2053229615001588 | |
 |
Abstract. Acidic hydrogen containing 2-isocyano-4-methylphenyl diphenylacetate, C22H17NO2, (I), was synthesized by the base-promoted reaction between 5-methylbenzoxazole and diphenylacetyl chloride. Achiral (I) crystallizes in the chiral P212121 space group. The C[triple bond]N bond length is 1.164 (2) Å and the angle between the OCO and 2-isocyano-4-methylphenyl planes is 69.10 (16)°. Molecules are linked via C=O…Hphenyl and bifurcated N[triple bond]C…Hphenyl/N[triple bond]C…Hmethine hydrogen bonds, forming one-dimensional arrays. | |
| 11 | J.J. Medvedev, O.S. Galkina, A.A. Klinkova, D.S. Giera, L. Hennig, C. Schneider, V.A. Nikolaev, “Domino [4 + 1]-annulation of α,β-unsaturated δ-amino esters with Rh(II)–carbenoids – a new approach towards multi-functionalized N-aryl pyrrolidines”, Org. Biomol. Chem. 2015, 13, 2640-2651 DOI:10.1039/C4OB02454K | |
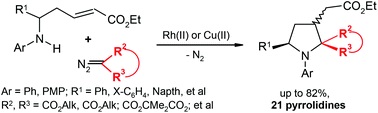 |
Abstract. Catalytic decomposition of diazomalonates and other diazoesters using Rh(II)- and Cu(II)-complexes in the presence of α,β-unsaturated δ-(N-aryl)amino esters gives rise to the formation of multi-functionalized pyrrolidines with yields of up to 82%. The reaction apparently occurs as a domino process involving the initial N-ylide formation followed by intramolecular Michael addition to the conjugated system of amino esters to afford the pyrrolidine heterocycle. The whole process can also be classified as a [4 + 1]-annulation of the δ-amino α,β-unsaturated ester with the carbenoid intermediate. | |
| 12 | N.V. Rostovskii, M.S. Novikov, A.F. Khlebnikov, G.L. Starova, M.S. Avdontseva, “Azirinium ylides from α-diazoketones and 2H-azirines on the route to 2H-1,4-oxazines: three-membered ring opening vs 1,5-cyclization”, Bellstein J. Org. Chem. 2015, 11, 302-312 DOI:10.3762/bjoc.11.35 | |
 |
Abstract. Strained azirinium ylides derived from 2H-azirines and α-diazoketones under Rh(II)-catalysis can undergo either irreversible ring opening across the N–C2 bond to 2-azabuta-1,3-dienes that further cyclize to 2H-1,4-oxazines or reversibly undergo a 1,5-cyclization to dihydroazireno[2,1-b]oxazoles. Dihydroazireno[2,1-b]oxazoles derived from 3-aryl-2H-azirines and 3-diazoacetylacetone or ethyl diazoacetoacetate are able to cycloadd to acetyl(methyl)ketene generated from 3-diazoacetylacetone under Rh(II) catalysis to give 4,6-dioxa-1-azabicyclo[3.2.1]oct-2-ene and/or 5,7-dioxa-1-azabicyclo[4.3.1]deca-3,8-diene-2-one derivatives. According to DFT calculations (B3LYP/6-31+G(d,p)), the cycloaddition can involve two modes of nucleophilic attack of the dihydroazireno[2,1-b]oxazole intermediate on acetyl(methyl)ketene followed by aziridine ring opening into atropoisomeric oxazolium betaines and cyclization. Azirinium ylides generated from 2,3-di- and 2,2,3-triaryl-substituted azirines give rise to only 2-azabuta-1,3-dienes and/or 2H-1,4-oxazines. | |
| 13 | K.S. Kisel, G. Linti, G.L. Starova, V.V. Sizov, A.S. Melnikov, A.P. Pushkarev, M.N. Bochkarev, E.V. Grachova, S.P. Tunik, “Syntheses, Structures, and Photophysical Properties of Eu and Lu Diketonates with a Neutral Polydentate Imidazolylmethanamine Ligand”, Eur. J. Inorg. Chem. 2015, 10, 1734-1743 DOI:10.1002/ejic.201403186 | |
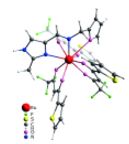 |
Abstract. The Schiff base NNO ligand 1-(furan-2-yl)-N-[(2-methyl-1H-imidazol-4-yl)methylene]methanamine was synthesized and structurally characterized by XRD crystallography, mass spectrometry, and NMR spectroscopy. Quantum-chemical calculations revealed conformational flexibility of the ligand backbone to give two different conformations with nearly equal ground-state energies. The orientation of two nitrogen atoms and the oxygen atom in one conformation is a good fit for the NNO tridentate coordination mode, whereas the other would allow the NN coordination mode only. Two lanthanide complexes [Ln(tta)3(NNO)] (Ln = EuIII and LuIII; tta = thenoyltrifluoroacetone) were prepared and studied spectroscopically. The structures of the complexes were optimized by the DFT approach. The NNO ligand in the Eu complex displays tridentate NNO coordination, whereas the ligand is only NN-coordinated in the Lu complex. The Eu complex shows bright red metal-centered phosphorescence under excitation into the ligand (π–π*) absorption bands with a quantum yield of ca. 80 % and a lifetime of 580 μs. A mechanism for energy transfer between the ligands and metal centers was suggested and confirmed by DFT and time-dependent DFT (TDDFT) studies. An organic light-emitting diode (OLED) device based on the Eu complex incorporated into a poly(9-vinylcarbazole) (PVK) matrix was prepared. A study of the characteristics of the device revealed electrochromism of the system owing to variations in the efficacy of metal-centered and matrix emission at different strengths of applied electric field. | |
| 14 | M.M. Efremova, A.P. Molchanov, A.V. Stepakov, R.R. Kostikov, V.S. Shcherbakova, A.V. Ivanov, “A highly efficient [3+2] cycloaddition of nitrile oxides and azomethine imines to N-vinylpyrroles”, Tetrahedron 2015, 71, 2071-2078 DOI:10.1016/j.tet.2015.02.058 | |
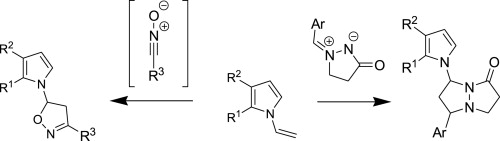 |
Abstract. 1,3-Dipolar cycloaddition of various nitrile oxides to substituted N-vinylpyrroles proceed with high efficiency and selectivity with the formation of single isomer of 5-pyrrolyl-substituted isoxazoline. The reaction of N-vinylpyrroles with cyclic azomethine imines occurs regioselectively affording 7-(pyrrol-1-yl)- substituted pyrazolo[1,2-a]pyrazolones as a mixture of two diastereomers. | |
| 15 | M.S. Ledovskaya, A.P. Molchanov, V.M. Boitsov, R.R. Kostikov, A.V. Stepakov, “An efficient synthesis of substituted isoxazolopyrroloisoquinolines via diastereoselective N-acyliminium ion cyclization”, Tetrahedron 2015, 71, 1952-1958 DOI:10.1016/j.tet.2015.02.031 | |
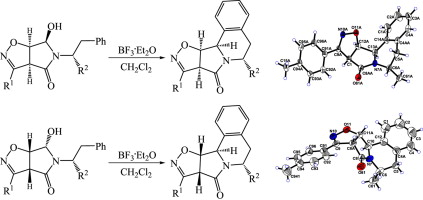 |
Abstract. A simple and efficient strategy was developed for the synthesis of fused pyrrolo[2,1-a]isoquinoline ring systems. The 5- and 6-substituted isoxazolopyrroloisoquinolines were readily prepared via diastereoselective N-acyliminium ion cyclization of 5-(1-R(or 2-R)-substituted-2-phenylethyl)-6-hydroxytetrahydro-4H-pyrrolo[3,4-d]isoxazol-4-ones derived from the corresponding bicyclic dihydroisoxazoles. | |
| 16 | A. Stepakov, S. Galkina, D. Bogomaz, E. Gaginskaya, A. Saifitdinova, “Modified Synthesis of 6-carboxyfluorescein (6-FAM): Application to Probe Labeling for Conventional Cytogenetics”, British J. App. Sci. Technol. 2015, 7, 423-428 DOI:10.9734/BJAST/2015/15991 | |
 |
Abstract. Aims: Fluorescent in situ hybridization (FISH), the routine technique of molecular cytogenetics, is widely used to detect and localize the presence of specific nucleic acids sequences in chromosomes, cell nucleus space, cells and tissue samples through the use of highly complementary probes to targets sequence. Expansion of FISH method application for research purposes and medical diagnostics requires efficient and low-cost production of labeled nucleic acid probes. Methodology and Results: We developed an effective method of fluorescein hydroxyalkyl carboxamides synthesis. This modification of the basic protocol of 6-carboxyfluorescein (6-FAM) synthesis enabled the production of highly reactive conjugate perfectly suitable for terminal labeling of newly generated oligonucleotides. Efficiency of 6-FAM labeled oligonucleotides obtained by the use of modified protocol has been proved for conventional cytogenetics. Conclusion: The suggested procedure of 6-FAM labeled oligonucleotides synthesis allows obtaining the high yield of directly labeled FISH probes. The introduction of this method into practice of cytogenetic studies will improve their efficiency and reduce the cost of an examination. |
|
| 17 | T.V. Serebryanskaya, A.S. Novikov, P.V. Gushchin, A.A. Zolotarev, V.V. Gurzhiy, V.Yu. Kukushkin, “Coupling of platinated triguanides with platinumactivated nitriles as a novel strategy for generation of dimetallic systems”, Dalton Trans. 2015, 44, 6003-6011 DOI:10.1039/c4dt03870c | |
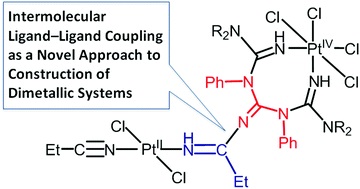 |
Abstract. One of two PtIV-activated propanenitriles in trans-[PtCl4(EtCN)2] is involved in platinum(IV)-mediated nitrile–imine coupling with the platinum(II)-based metallacycles [PtCl2{NH[double bond, length as m-dash]C(NR2)N(Ph)C([double bond, length as m-dash]NH)N(Ph)C(NR2)[double bond, length as m-dash]NH}] [R2 = Me2 (1a), C5H10 (1b)] yielding diplatinum products, whose structures depend on molar ratios between the reactants. At a 1 : 1 ratio, the mixed-valence platinum(II)/platinum(IV) species [PtCl4{NH[double bond, length as m-dash]C(NR2)N(Ph)C{[double bond, length as m-dash][(N(Et)C[double bond, length as m-dash]NH)PtCl2(EtCN)]}N(Ph)C(NR2)[double bond, length as m-dash]NH}] [R2 = Me2 (2a), (CH2)5 (2b)] were generated, whereas at a 1 : 2 ratio the dinuclear platinum(II)/platinum(II) complexes [PtCl2{NH[double bond, length as m-dash]C(NR2)N(Ph)C{[double bond, length as m-dash][(N(Et)C[double bond, length as m-dash]NH)PtCl2(EtCN)]}N(Ph)C(NR2)[double bond, length as m-dash]NH}] [R2 = Me2 (3a), (CH2)5 (3b)] were obtained. In contrast to the nitrile–imine coupling observed for the platinum(IV) dinitrile complex, the reaction between the platinum(II) congener trans-[PtCl2(EtCN)2] and any one of 1a,b gives exclusively the substituted dimetallic platinum(II)/platinum(II) products [PtCl2{NH[double bond, length as m-dash]C(NR2)N(Ph)C{[double bond, length as m-dash][(NH)PtCl2(EtCN)]}N(Ph)C(NR2)[double bond, length as m-dash]NH}] [R2 = Me2 (6a), (CH2)5 (6b)] featuring platinum-containing guanidine 1 as one of the ligands. Complexes 2a,b, 3a,b, and 6a,b were characterized by elemental analyses (C, H, N), HRESI-MS, IR, 1H NMR spectroscopy, and DTA/TG. The molecular and crystal structure of 2a·2CDCl3 was additionally studied by single-crystal X-ray diffraction. Complexes 2a,b undergo further redox transformation in solutions, and single crystals of [PtCl2{NH[double bond, length as m-dash]C(NMe2)N(Ph)C{[double bond, length as m-dash][(N(Et)C[double bond, length as m-dash]NH)PtCl2(MeCN)]}N(Ph)C(NMe2)[double bond, length as m-dash]NH}]·2CH2Cl2 (3′a·2CH2Cl2) were obtained from 2a in a CH2Cl2–MeCN–C2H4Cl2 mixture and studied by X-ray crystallography. The driving forces for the generation of diplatinum products 2 and 3 were elucidated based on a quantum-chemical study. | |
| 18 | В.А. Федюкевич, С. А. Кубышкин, А. А. Блохин, С.М. Сухаржевский, Н.В. Воробьев-Десятовский, «Использование ионообменных смол для борьбы с явлением прег-роббинга золота в процессе цианидного выщелачивания», Журнал Прикладной Химии 2015, 88, 246-255 | |
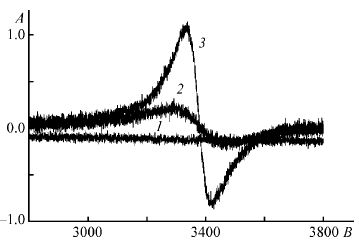 |
Abstract. Изучена возможность применения сильноосновных анионитов марки MINIX и слабоосновных анионитов марки Purogold S992 в технологическом процессе High Temperature Caustic Conditioning (HiTeCC) для уменьшения потерь золота от адсорбции на природных органических углеродсодержащих веществах (явления прег-роббинга) в ходе цианидного выщелачивания. Показано, что сильноосновный анионит MINIX обладает более высоким сродством к аниону [Au(CN)2 ]– по сравнению с микропористым активированным углем WSC-207C-GR. Следствием этого является перенос адсорбированного дицианоауратного(I) аниона с насыщенного угля на ионообменную смолу. Рассмотрены причины связывания дицианоауратного(I) иона с активными центрами активированных углей, основанные на существовании в составе адсорбента карбеноподобных атомов углерода. Наличие в составе активированного угля парамагнитных центров, принадлежащих атомам углерода, показано методом ЭПР. Предложенная концепция использована для уменьшения потерь золота, связанного с частичным окислением металлического золота при автоклавной переработке пирит и арсенопиритсодержащих концентратов. |
|
| 19 | A.P. Molchanov, Yu.V. Malinina, R.R. Kostikov, A.V. Stepakov, “Regioselective Cycloaddition of C,N-Diarylnitrones to Arylallenes and of N-Aryl-C-carbamoylnitrones to Methyl Buta-2,3-dienoate”, Russ. J. Org. Chem. 2015, 51, 368-372 DOI:10.1134/S1070428015030136 | |
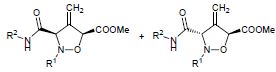 |
Abstract. Cycloadditions of C,N-diarylnitrones to non-activated arylallenes and of N-aryl-C-carbamoylnitrones to methyl buta-2,3-dienoate regioselectively afforded mixtures of diastereoisomeric substituted 4-methylideneisoxazolidines. | |
| 20 | A.V. Stepakov, A.G. Larina, A.P. Molchanov, “Isomerization of Dimethylenecyclopropanes in Benzofulvenes in the Presence of Lewis Acids”, Russ. J. Org. Chem. 2015, 51, 210-213 DOI:10.1134/S1070428015020128 | |
 |
Abstract. 1,1-Diaryl-2-(1-methylethylidene)-3-methylenecyclopropanes in the presence of Lewis acid isomerize in benzofulvene derivatives. | |
| 21 | T. Kozlecki, P.M. Tolstoy, A. Kwocz, M.A. Vovk, A. Kochel, I. Polowczyka, P.Yu. Tretyakov, A. Filarowski, “Conformational state of β-hydroxynaphthylamides and barriers for the rotation of the amide group”, Spectrochimica Acta A 2015, 149, 254-262 DOI:10.1016/j.saa.2015.04.052 | |
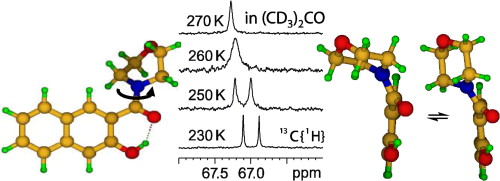 |
Abstract. Three β-hydroxynaphthylamides (morpholine, pyrrolidine and dimethylamine derivatives) have been synthesized and their conformational state was analyzed by NMR, X-ray and DFT calculations. In aprotic solution the molecules contain intramolecular OHO hydrogen bonds, which change into intermolecular ones in solid state. The energy barriers for the amide group rotation around the CN bond were estimated from the line shape analysis of 1H and 13C NMR signals. A tentative correlation between the barrier height and the strength of OHO bond was proposed. Calculations of the potential energy profiles for the rotations around CC and CN bonds were done. In case of morpholine derivative experimental indications of additional dynamics: chair–chair ‘ring flip’ in combination with the twisting around CC bond were obtained and confirmed by quantum chemistry calculations. | |
| 22 | Е.А. Катленок, А.А. Золотарев, А.Ю. Иванов, С.Н. Смирнов, Р.И. Байчурин, К.П. Балашев, “Cтроение, оптические и электрохимические свойства биядерных платинированных комплексов 2-фенилбензотиазола с мостиковыми 2-меркаптопроизводными тиазолина, 1-метилимидазола и пиримидина”, Коорд. Химия 2015, 41(6), 357-364 DOI:10.7868/S0132344X15060031 | |
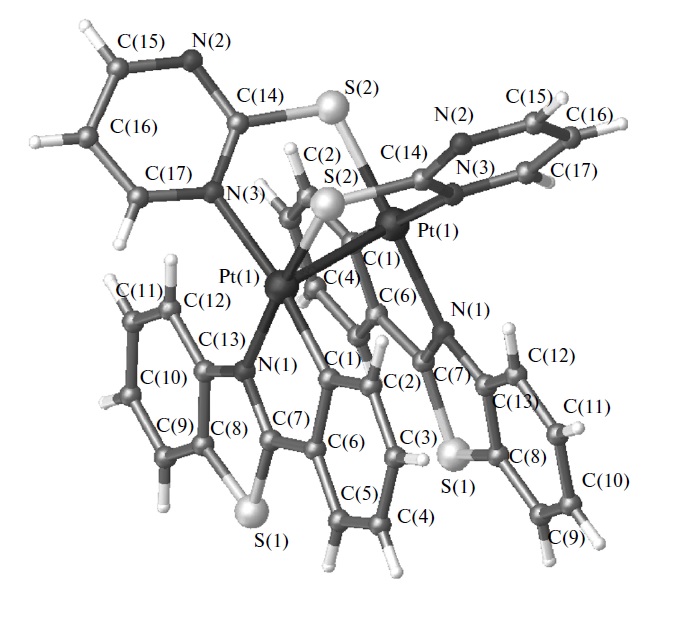 |
Abstract. Методами РСА и спектроскопии ЯМР 1, 13, 195Pt исследовано строение комплексов [Pt(Bt)( -(N^S))]2 с платинированным 2-фенилбензотиазолом (Bt) и мостиковыми 2-меркаптопроизводными (N^S) тиазолина, 1-метилимидазола и пиримидина в кристаллическом состоянии и растворе CDCl3. Показано цис-N(Bt),S(N^S)-строение комплексов [Pt(Bt)(u-(N^S))]2 с химической связью PtPt 2.892.93 A и антисимметричным положением двух циклоплатинированных и двух мостиковых лигандов (CCDC № 1001204 (I), 1027021 (II), 993566 (III)). Длинноволновые полосы поглощения (410540 нм), полосы фосфоресценции с маx λ= 685 нм, процессы двухэлектронного электровосстановления c Ep = (2.002.27) В и окисления c Ep = 0.070.20 В отнесены к фото- и электростимулированным процессам с участием НСМО и ВЗМО, преимущественно локализованных на σ*-орбиталях {Pt(Bt)} и σ*-орбиталях химической связи металлметалл. | |
| 23 | S.N. Britvin, A. Lotnyk, “Water-Soluble Phosphine Capable of Dissolving Elemental Gold: The Missing Link between 1,3,5-Triaza-7-phosphaadamantane (PTA) and Verkade’s Ephemeral Ligand”, J. Am. Chem. Soc. 2015, 137, 5526-5535 DOI:10.1021/jacs.5b01851 | |
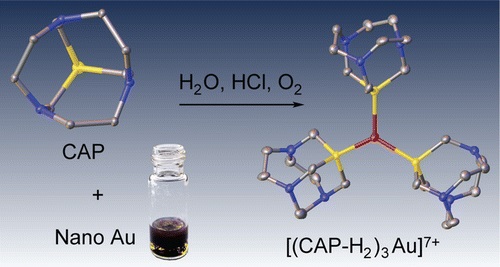 |
Abstract. We herein describe a tricyclic phosphine with previously unreported tris(homoadamantane) cage architecture. That water-soluble, air- and thermally stable ligand, 1,4,7-triaza-9-phosphatricyclo[5.3.2.14,9]tridecane (hereinafter referred to as CAP) exhibits unusual chemical behavior toward gold and gold compounds: it readily reduces Au(III) to Au(0), promotes oxidative dissolution of nanocrystalline gold(0) with the formation of water-soluble trigonal CAP–Au(I) complexes, and displaces cyanide from [Au(CN)2]− affording triangular [Au(CAP)3]+ cation. From the stereochemical point of view, CAP can be regarded as an intermediate between 1,3,5-triaza-7-phosphaadamantane (PTA) and very unstable aminophosphine synthesized by Verkade’s group: hexahydro-2a,4a,6a-triaza-6b-phosphacyclopenta[cd]pentalene. The chemical properties of CAP are likely related to its anomalous stereoelectronic profile: combination of strong electron-donating power (Tolman’s electronic parameter 2056.8 cm–1) with the low steric demand (cone angle of 109°). CAP can be considered as macrocyclic counterpart of PTA with the electron-donating power approaching that of strongest known phosphine electron donors such as P(t-Bu)3 and PCy3. Therefore, CAP as sterically undemanding and electron-rich ligand populates the empty field on the stereoelectronic map of phosphine ligands: the niche between the classic tertiary phosphines and the sterically undemanding aminophosphines. | |
| 24 | P.R. Golubev, A.S. Pankova, M.A. Kuznetsov, “Regioselective Transition-Metal-Free Synthesis of 2‑(Trimethylsilylmethylene)pyrrol-3-ones by Thermal Cyclization of Acetylenic Enamines”, J. Org. Chem. 2015, 80, 4545-4552 DOI:10.1021/acs.joc.5b00398 | |
 |
Abstract. Acetylenic enamines generated in situ from readily available enynones and primary amines undergo thermal cyclization in diphenyl ether providing easy access to 4-aryl-2-(trimethylsilylmethylene)-1,2-dihydro-3H-pyrrol-3-ones. This reaction is inherently versatile, allowing for variations of substituents in both enynone and amine. Full regioselectivity along with short reaction time (1–2 h) and simple workup afford single products in good to excellent isolated yields. Fluorescent properties of the obtained compounds were studied. | |
| 25 | A.S. Pankova, A.Yu. Stukalov, M.A. Kuznetsov, “Synthesis of 2‑(Hetero)aryl-5-(trimethylsilylethynyl)oxazoles from 2 (Hetero)arylacrylic Acids”, Org. Lett. 2015, 17, 1826-1829 DOI:10.1021/acs.orglett.5b00009 | |
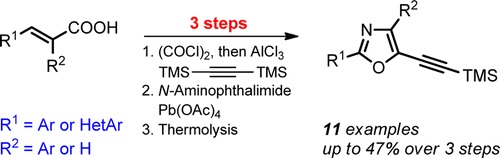 |
Abstract. A three-step method for the synthesis of 2-(hetero)aryl-5-(trimethylsilylethynyl)oxazoles is described. Easily accessible bis(trimethylsilyl)acetylene and acrylic acid derivatives are used as starting materials for the preparation of mono- and disubstituted 5-(trimethylsilyl)pent-1-en-4-yn-3-ones. Oxidative phthalimidoaziridination of these enynones provides the key 2-acyl-1-phthalimidoaziridines that are further utilized in the thermal expansion of the three-membered ring to furnish the target functionalizable oxazoles. | |
| 26 | R.O. Iakovenko, A.N. Kazakova, V.M. Muzalevskiy, A.Yu. Ivanov, I.A. Boyarskaya, A. Chicca, V. Petrucci, J. Gertsch, M. Krasavin, G.L. Starova, A.A. Zolotarev, M.S. Avdontceva, V.G. Nenajdenko, A.V. Vasilyev, “Reactions of CF3-enones with arenes under superelectrophilic activation: a pathway to trans-1,3-diaryl-1-CF3-indanes, new cannabinoid receptor ligands”, Org. Biomol. Chem. 2015, 13, 8827-8842 DOI:10.1039/c5ob01072a | |
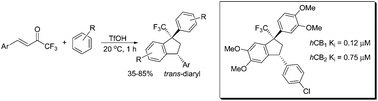 |
Abstract. 4-Aryl-1,1,1-trifluorobut-3-en-2-ones ArCH[double bond, length as m-dash]CHCOCF3 (CF3-enones) react with arenes in excess of Brønsted superacids (TfOH, FSO3H) to give, stereoselectively, trans-1,3-diaryl-1-trifluoromethyl indanes in 35–85% yields. The reaction intermediates, the O-protonated ArCH[double bond, length as m-dash]CHC(OH+)CF3 and the O,C-diprotonated ArHC+CH2C(OH+)CF3 species, have been studied by means of 1H, 13C, 19F NMR, and DFT calculations. Both types of the cations may participate in the reaction, depending on their electrophilicity and electron-donating properties of the arenes. The formation of CF3-indanes is a result of cascade reaction of protonated CF3-enones to form chemo-, regio- and stereoselectively three new C–C bonds. The obtained trans-1,3-diaryl-1-trifluoromethyl indanes were investigated as potential ligands for cannabinoid receptors CB1 and CB2 types. The most potent compound showed sub-micromolar affinity for both receptor subtypes with a 6-fold selectivity toward the CB2 receptor with no appreciable cytotoxicity toward SHSY5Y cells. | |
| 27 | M.E. Chizhova, O.Yu. Bakulina, A.Yu. Ivanov, P.S. Lobanov, D.V. Dar’in, “Facile synthesis of pyrido[2,3-d]pyrimidines via cyclocondensation of 4,6-dichloro-2-methylsulfanylpyrimidine-5-carbaldehyde with β-substituted β-aminoacrylic esters”, Tetrahedron 2015, 71, 6196-6203 DOI:10.1016/j.tet.2015.06.085 | |
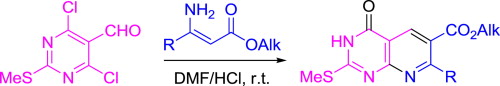 |
Abstract. A new facile synthesis of pyrido[2,3-d]pyrimidin-4-ones via cyclocondensation of 4,6-dichloro-2-methylsulfanylpyrimidine-5-carbaldehyde with β-alkyl and β-aryl-β-aminoacrylic esters followed by hydrolysis of chlorine atom at position 4 of pyridopyrimidine ring has been developed. The cyclocondensation was found to be accelerated by acid. | |
| 28 | D.V. Dar’in, А.Yu. Ivanov, P.S. Lobanov, “Cyclocondensation of Ethyl (imidazolidine-2-ylidene)acetate with Aromatic Esters Bearing Labile Halogen in ortho-Position”, J. Heterocyclic Chem. 2015, 52, 1192-1194 DOI:10.1002/jhet.2229 | |
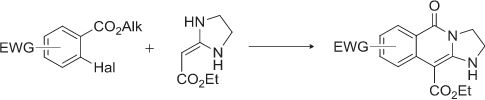 |
Abstract. Cyclocondensation of ethyl (imidazolidine-2-ylidene)acetate with aromatic esters bearing labile halogen in ortho-position leads to fused heterocycles, which is formed by substitution of halogen atom with α-carbon atom of cyclic ketene aminal and binding of nitrogen atom with carbonyl carbon atom of aromatic ester. | |
| 29 | S. Lednev, A. Sirick, E. Pliss, A. Rusakov, N. Shvyrkova, A. Ivanov. “Influence of solvation on the kinetics of methyl methacrylate oxidation inhibited by aromatic amines”, Reac. Kinet. Mech. Cat. 2015, 116, 43-50 DOI:10.1007/s11144-015-0881-9 | |
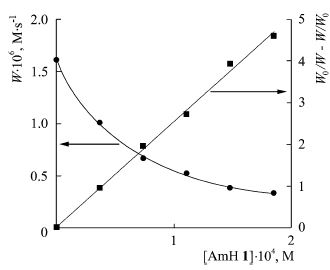 |
Abstract. The kinetics of methyl methacrylate oxidation inhibited by aromatic amines in media of different polarities was studied. The complexing parameters in systems “oxidation substrate—aromatic amine—solvent” using IR and NMR spectroscopy were found. The correlations between inhibited oxidation rate constant and the polarity of the medium were obtained using Kirkwood–Onsager equation. | |
| 30 | A.S. Pankova, M.V. Sorokina, М.А. Кuznetsov, “Thermal rearrangement of 2,3-diaryl-1-phthalimidoaziridines”, Tetrahedron Lett. 2015, 56, 5381-5385 DOI:10.1016/j.tetlet.2015.07.093 | |
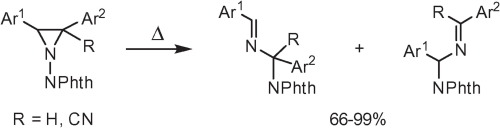 |
Abstract. 2,3-Diaryl-1-phthalimidoaziridines and 2,3-diaryl-1-phthalimidoaziridine-2-carbonitriles were found to readily undergo thermal rearrangement into imines via 1,2-migration of the phthalimido group and accompanying C–C bond cleavage. Isomerization proceeds regioselectively with preferable migration to the electron-deficient carbon atom. Interestingly, this reaction was found to predominate even in the presence of dipolarophiles. | |
| 31 | A. Antonov, A. Pozharskii, V. Ozeranskii, A. Filarowski, K. Suponitsky, P. Tolstoy, “Ring Lithiation of 1,8-Bis(dimethylamino)naphthalene: Another Side of the ‘Proton Sponge Coin’”, Dalton Trans. 2015, 44, 17756-17766 DOI:10.1039/C5DT02482J | |
 |
Abstract. It has been found that 1,8-bis(dimethylamino)naphthalene (DMAN), unlike N,N-dimethylaniline, undergoes ring metallation in the n-BuLi–TMEDA–Et2O system with a low selectivity and in poor total yields. The situation is significantly improved in the t-BuLi–TMEDA–n-hexane system when 3- and 4-lithium derivatives become the only reaction products obtained in good yields. The formation of 3-Li-DMAN is especially desired since no method of direct meta-functionalization of DMAN is known to date. The relative stability and structure of DMAN lithium derivatives have been examined with the help of X-ray and multinuclear NMR measurements as well as DFT calculations. | |
| 32 | A.N. Kazakova, R.O. Iakovenko, I.A. Boyarskaya, V.G. Nenajdenko, A.V. Vasilyev, “Acid-Promoted Reaction of Trifluoromethylated Allyl Alcohols with Arenes. Stereoselective Synthesis of CF3‑Alkenes and CF3‑Indanes”, J Org. Chem. 2015, 80, 9506-9517 DOI:10.1021/acs.joc.5b01398 | |
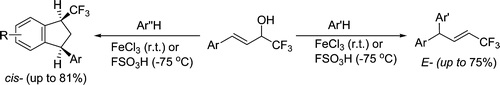 |
Abstract. Reaction of 4-aryl-1,1,1-trifluorobut-3-en-2-ols [CF3-allyl alcohols, ArCH═CHCH(OH)CF3] with arenes under activation with anhydrous FeCl3 or FSO3H was studied. We found that the transformation led to trifluoromethylated alkenes [Ar(Ar′)CHCH═CHCF3] or 1-trifluoromethylated indanes (CF3-indanes). The formation of these two types of reaction products strongly depends on the nucleophilicity of the starting arene and the electrophilicity of cationic intermediates generated from CF3-allyl alcohols under reaction conditions. Benzene, anisole, veratrole, and ortho-xylene lead exclusively to CF3-alkenes with an E-configuration. More π-donating polymethylated arenes (pseudocumene, mesitylene) afford only CF3-indanes with a predominantly cis-orientation of substituents at positions 1 and 3 of the indane ring. Meta- and para-xylenes show an intermediate behavior; they may form both CF3-alkenes and/or CF3-indanes. The mechanisms of the investigated transformations are discussed. | |
| 33 | D.I. Nilov, A.V. Vasilyev, “One-pot tandem hydrophenylation and ionic hydrogenation of 3-phenylpropynoic acid derivatives under superelectrophilic activation”, Tetrahedron Lett. 2015, 56, 5714-5717 DOI:10.1016/j.tetlet.2015.09.026 | |
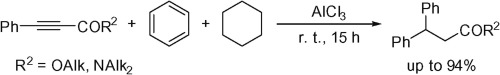 |
Abstract. The reactions of esters and amides of 3-phenylpropynoic acid with strong Lewis acids AlX3 (X = Cl, Br) or conjugate Brønsted–Lewis superacids HX-AlX3 (X = Cl, Br) in benzene and cyclohexane at room temperature afforded 3,3-diphenylpropanoic acid derivatives in up to 94% yield. This tandem reaction of the acetylene bond proceeded by hydrophenylation followed by ionic hydrogenation. | |
| 34 | Э.И. Евстигнеев, О.С. Юзихин, А.А. Гуринов, А.Ю. Иванов, Т.О. Артамонова, М.А. Ходорковский, Е.А. Бессонова, А.В. Васильев, «Химическое строение и физико-химические свойства окисленного гидролизного лигнина», Журнал прикладной химии 2015, 88(8), 1175-118 | |
| 35 | S.O. Rabdano, A.V. Donets, M.A. Vovk, D.Michel, V.I. Chizhik, ““Hydration Shells” of CH2 Groups of ω‑Amino Acids as Studied by Deuteron NMR Relaxation”, J. Phys. Chem. B 2015, 119, 13358-13366 DOI:10.1021/acs.jpcb.5b06584 | |
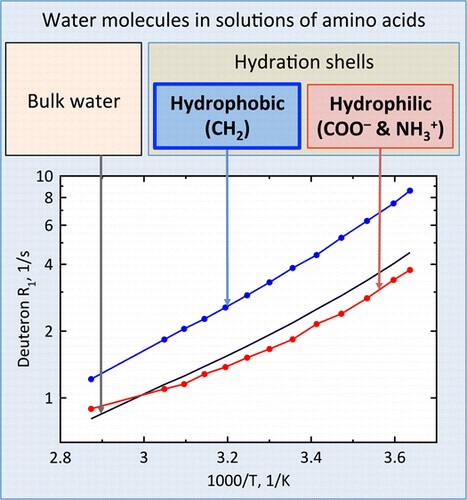 |
Abstract. Hydration phenomena play a very important role in various processes, in particular in biological systems. Water molecules in aqueous solutions of organic compounds can be distributed among the following substructures: (i) hydration shells of hydrophilic functional groups of molecules, (ii) water in the environment of nonpolar moieties, and (iii) bulk water. Up to now, the values of hydration parameters suggested for the description of various solutions of organic compounds were not thoroughly analyzed in the aspect of the consideration of the total molecular composition. The temperature and concentration dependences of relaxation rates of water deuterons were studied in a wide range of concentration and temperature in aqueous (D2O) solutions of a set of ω-amino acids. Assuming the coordination number of the CH2 group equal to 7, which was determined from quantum-chemical calculations, it was found that the rotational correlation times of water molecules near the methylene group is 1.5–2 times greater than one for pure water. The average rotational mobility of water molecules in the hydration shells of hydrophilic groups of ω-amino acids is a bit slower than that in pure solvent at temperatures higher that 60 °C, but at lower temperatures, it is 0.8–1.0 of values of correlation times for bulk water. The technique suggested provides the basis for the characterization of different hydrophobic and hydrophilic species in the convenient terms of the rotational correlation times for the nearest water molecules. | |
| 36 | V.V. Suslonov, N. Martines-Rodriges, L.R. Mingabudinova, M.G. Osmolovskii, O.M. Osmolovskaya, “Features of reduction of nickel, cobalt, and copper ions in polyol processes”, Russ. J. Org. Chem. 2015, 85, 1768-1770 DOI:10.1134/S1070363215070324 | |
| 37 | A.A. Melekhova, A.S. Novikov, K.V. Luzyanin, N.A. Bokach, G.L. Starova, V.V. Gurzhiy, V.Yu. Kukushkin, “Tris-isocyanide copper(I) complexes: Synthetic, structural, and theoretical study”, Inorg. Chim. Acta 2015, 434, 31-36 DOI:10.1016/j.ica.2015.05.002 | |
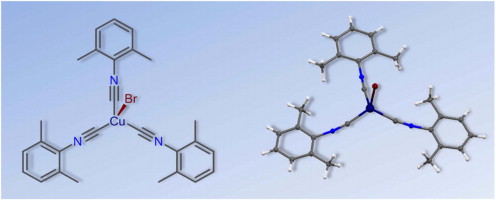 |
Abstract. The reaction of CuBr with 3 equiv of CNR in CHCl3 at RT furnished the pure tris-isocyanide complexes [CuBr(CNR)3] [R = 2,6-Me2C6H3 (Xyl) (1), 2-Cl-6-MeC6H3 (2), 2-Naphthyl (3), C6H11 (Cy) (4)] in 85–99% isolated yield. Compounds 1–4 were characterized by elemental analyses (C, H, N), high resolution ESI+-MS, IR, 1H and 13C{1H} NMR spectroscopic techniques, and by single-crystal X-ray diffraction for 1 and 2. Coordination polyhedra of these two complexes are intermediate between tetrahedral and trigonal pyramidal with three isocyanide and one bromide ligands; the fragment M–C–N is bent [171.2(2)–175.6(2)°]. The influence of crystal packing effects on the geometric parameters of 1 as well as nature of the coordination Cu–C bonds and bond order of the formal triple Ctriple bond; length of mdashN bonds in this compound were also studied theoretically at the DFT level of theory. The crystal packing effects noticeably affect the values of the Cu–C–N angles and lead to a decrease of these angles approximately 10° relative to the gas phase geometries. Results of the AIM, NBO, and CDA analyses reveal that the Cu–C coordination bonds are of the electrostatic nature. Electrostatic interaction, unlike the covalent one, has no orientation, and it is the reason of the curved shape of the metal–ligand fragment. | |
| 38 | S.A. Timofeeva, M.A. Kinzhalov, V.P. Boyarskiy, T.M. Buslaeva, E.A. Valishina, M. Haukka, K.V. Luzyanin, V.Yu. Kukushkin, “Application of palladium complexes bearing acyclic amino(hydrazido)carbene ligands as catalysts for copper-free Sonogashira cross-coupling”, J. Catalysis 2015, 329, 449-456 DOI:10.1016/j.jcat.2015.06.001 | |
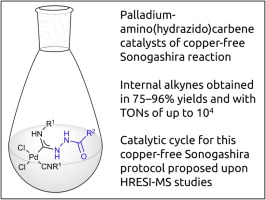 |
Abstract. Metal-mediated coupling of one isocyanide in cis-[PdCl2(CNR1)2] (R1 = C6H11 (Cy) 1, tBu 2, 2,6-Me2C6H3 (Xyl) 3, 2-Cl-6-MeC6H34) and various carbohydrazides R2CONHNH2 [R2 = Ph 5, 4-ClC6H46, 3-NO2C6H47, 4-NO2C6H48, 4-CH3C6H49, 3,4-(MeO)2C6H310, naphth-1-yl 11, fur-2-yl 12, 4-NO2C6H4CH213, Cy 14, 1-(4-fluorophenyl)-5-oxopyrrolidine-3-yl 15, (pyrrolidin-1-yl)C(O) 16, 3-(3,5-di-tert-butyl-4-hydroxyphenyl)propane-1-yl 17, EtNHC(O) 18] or sulfohydrazides R3SO2NHNH2 [R3 = Ph 19, 4-MeC6H420] led to a series of (hydrazido)(amino)carbene complexes cis-[PdCl2{C(NHNHX)double bond; length as m-dashN(H)R1}(CNR1)]; X = COR2, SO2R3 (21–48, isolated yields 60–96%). All prepared species were characterized by elemental analyses (C, H, N), HR ESI+-MS, IR, 1H and 13C{1H} NMR spectroscopy, and by a single-crystal X-ray diffraction for 38. Complexes 21–48 demonstrated excellent activity as catalysts in copper-free Sonogashira coupling of aryl iodides and a variety of aromatic terminal alkynes. Catalytic system runs in environmentally benign EtOH ensuring product yields of up to 75–96% and TONs of up to 104. Mechanism of the copper-free Sonogashira catalytic cycle involving 21–48 as catalysts was proposed upon identification of key intermediates using HRESI-mass. | |
| 39 | Yu.B. Koptelov, D.O. Antuganov, A.P. Molchanov, R.R. Kostikov, “Steric Hindrances to the Cycloaddition of (Z)-1-Arylmethylidene-5,5-dimethyl-3-oxopyrazolidin-1-ium-2-ides to N-Arylmaleimides”, Russ. J. Org. Chem. 2015, 7, 972-981 DOI:10.1134/S1070428015070143 | |
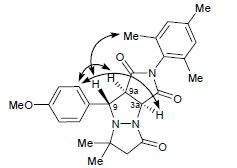 |
Abstract. Sterically hindered cycloaddition of (Z)-1-arylmethylidene-5,5-dimethyl-3-oxopyrazolidin-1-ium-2-ides to 4-mono- and 2,6-disubstituted N-arylmaleimides requires prolonged heating (40–60 h) at ~150–155°C and yields mixtures of diastereoisomeric cycloadducts. The observed diastereoselectivity is determined by both electronic and steric interactions, depending on the nature and position of substituents in the azomethine imine and maleimide. The reactions of (Z)-1-(2,6-dichlorobenzylidene)-5,5-dimethyl-3-oxopyrazolidin-1-ium-2-ide with 4-substituted N-arylmaleimides give mainly the corresponding cis adducts as a result of preferential exo attack by the dipolarophile, whereas trans adducts predominate in the cycloaddition of (Z)-1-(4-X-benzylidene)-5,5-dimethyl-3-oxopyrazolidin-1-ium-2-ide and (Z)-1-(2,6-dichlorobenzylidene)-5,5-dimethyl-3-oxopyrazolidin- 1-ium-2-ide to 2,6-disubstituted N-arylmaleimides. | |
| 40 | D.V. Kurandina, E.V. Eliseenkov, T.S. Khaibulova, A.A. Petrov, V.P. Boyarskiy, “Copper-catalyzed C-N bond cross-coupling of aryl halides and amines in water in the presence of ligand derived oxalyl dihydrazide: scope and limitation”, Tetrahedron 2015, 71, 7931-7937 DOI:10.1016/j.tet.2015.07.071 | |
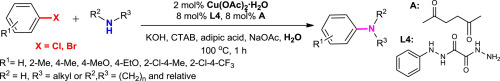 |
Abstract. An efficient and convenient method has been developed for the copper-catalyzed C–N bond cross-coupling of aryl bromides with electron-donor substituents and aliphatic amines in water. The new ligand system N-phenyloxalyl bishydrazide/hexane-2,5-dione has been shown to be considerably more efficient in the copper-catalyzed C–N bond cross-coupling reaction as compared to the ligands described in the literature and allowed decreasing of the catalyst amount (up to 2 mol %) to achieve acceptable yields of isolated products (46–84%). Acceptor substituted aryl bromides, aryl bromides with substituents in the ortho-position, and some aryl dichlorides can undergo the C–N cross-coupling under the developed conditions, but their reactivity is lower. | |
| 41 | D.S. Grosheva, V.A. Rassadin, V.V. Sokolov, “A Route to Benzo-Annelated δ-Sultams through Michael Cyclization”, Eur. J. Org. Chem. 2015, 6, 1355-1363 DOI:10.1002/ejoc.201403416 | |
 |
Abstract. A new approach to benzo-annelated δ-sultams containing an aryl–nitrogen bond is described. The method, which involves the intramolecular Michael cyclization of tert-butyl ortho-[N-(methoxycarbonylmethyl)sulfonylamino]cinnamates, allows the synthesis of secondary sultams as well as their tertiary analogues and bridged tricyclic derivatives efficiently and diastereoselectively. | |
| 42 | S.N. Britvin, S.A. Kashtanov, M.G. Krzhizhanovskaya, A.A. Gurinov, O.V. Glumov, S. Strekopytov, Yu.L. Kretser, A.N. Zaitsev, N.V. Chukanov, S.V. Krivovichev, “Perovskites with the Framework-Forming Xenon”, Angew. Chem. Int. Ed. 2015, 54, 1-6 DOI:10.1002/anie.201506690 | |
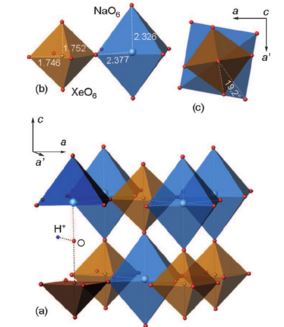 |
Abstract. The Group 18 elements (noble gases) were the last ones in the periodic system to have not been encountered in perovskite structures. We herein report the synthesis of a new group of double perovskites KM(XeNaO6) (M=Ca, Sr, Ba) containing framework-forming xenon. The structures of the new compounds, like other double perovskites, are built up of the alternating sequence of corner-sharing (XeO6) and (NaO6) octahedra arranged in a three-dimensional rocksalt order. The fact that xenon can be incorporated into the perovskite structure provides new insights into the problem of Xe depletion in the atmosphere. Since octahedrally coordinated XeVIII and SiIV exhibit close values of ionic radii (0.48 and 0.40 Å, respectively), one could assume that XeVIII can be incorporated into hyperbaric frameworks such as MgSiO3 perovskite. The ability of Xe to form stable inorganic frameworks can further extend the rich and still enigmatic chemistry of this noble gas. | |
| 43 | I.S. Ignatyev, T.A. Kochina, V.V. Avrorin, V.V. Gurzha, I.M. Fundamensky, “Molecular and crystal structure of 2-phenyl-2-hydro-6-methyl-1,3-dioxa-6-aza-2-silacyclooctane”, J. Molec. Struct. 2015, 1094, 169-173 DOI:10.1016/j.molstruc.2015.03.061 | |
 |
Abstract. The crystal structure of 2-phenyl-2-hydro-6-methyl-1,3-dioxa-6-aza-2-silacyclooctane [HPhSi(OCH2CH2)2NMe – phenylhydrosilocane (I)] is determined by single-crystal X-ray diffraction at 100 K. The unit cell consists of four molecules connected only by Van-der-Waals interactions. Each molecule has an eight-membered heterocycle with a phenyl group in the axial position. The Si⋯N transannular bond has a short (2.206 Å) interatomic distance which exceeds only this distance in ocanes with highly electronegative fluorine substituents at Si. Since there exist experimental data on the occurrence of different conformers of I in the liquid phase, the PES of the molecule was analyzed by DFT B3LYP and MP2 methods with the aug-cc-pVDZ basis set. The energy minimum belongs to the boat–chair conformation with the axial position of the phenyl group. Rotation of the phenyl ring around the SiC bond has a barrier ca. 1 kcal/mol. The conformer with the equatorial position of this group lies 6 kcal/mol higher. Interconversion of this conformers which was observed in experiment proceeds through the chair–chair configuration in which the Si⋯N transannular bond is absent and coordination at silicon is tetrahedral, rather than trigonal bipyramidal one observed in other conformers. | |
| 44 | Е.В. Абакумов, Ю.М. Фаттахова, «Структурный состав гуминовых веществ орнитогенных почв Антарктики по данным ядерного магнитного резонанса (13-С)», Русский орнитологический журнал, 2015, 1165, 2463-2466 | |
| 45 | A.P. Molchanov, E.V. Sirotkina, M.M. Efremova, R.R. Kostikov, A.V. Ivanov, V.S. Shcherbakova, “Regio- and Stereoive Cycloaddition of Nitrones to 1-Vinyl-4,5-dihydro-1Н-benzo[g]indole”, Russ. J. Org. Chem. 2015, 51, 640-643 DOI:10.1134/S1070428015050097 | |
 |
Abstract. Nitrones cycloaddition to 1-vinyl-4,5-dihydro-1Н-benzo[g]indole proceeds regio- and steroselectively affording a single diastereomer of 5-(hetaryl-substituted)isoxazolidine. | |
| 46 | Yu.B. Koptelov, A.P. Molchanov, R.R. Kostikov, “Regio- and Diastereoive Cycloaddition of Stable Cyclic Azomethine Imines to N-Arylitaconic Imides”, Russ. J. Org. Chem. 2015, 8, 1134-1143 DOI:10.1134/S1070428015080126 | |
 |
Abstract. 1,3-Dipolar cycloaddition of stable N,N′- and C,N-cyclic azomethine imines based respectively on pyrazolidin-3-one and 3,4-dihydroisoquinoline to N-arylitaconimides proceeds strictly regioselectively resulting in spiro-joint heterocycles in good yields. The regioselectivity of the addition of N,N′-cyclic azomethine imines is reversed with respect to that of C,N-cyclic azomethine imines. The diastereoselectivity of the cycloaddition depends apparently on the substituents at the central nitrogen atom in the azomethine imine fragment. | |
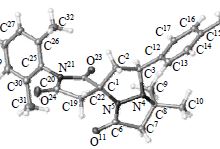
| 47 | A.D. Lisakova, D.S. Ryabukhin, R.E. Trifonov, V.A. Ostrovskii, A.V. Vasilyev, “of 2-Methyl-5-vinyl-2H-tetrazole with Iodoarenes”, Russ. J. Org. Chem. 2015, 2, 290-291 DOI:10.1134/S1070428015020293 | |
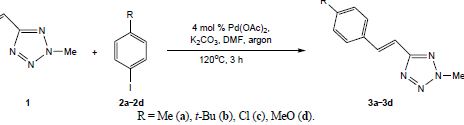 |
||
| 48 | N.I. Svintsitskaya, А.V. Dogadina, R.Е. Trifonov, “Synthesis of tetrazolyl-containing C-phosphonoacetamidines”, Synlett 2015, 26, A-D DOI:10.1055/s-0035-1560505 | |
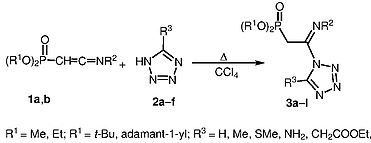 |
Abstract. Addition of 5-substituted tetrazoles to dialkyl 2-(N-tert-butyl- or N-adamantyl)iminovinylphosphonates proceeds regioselectively to yield novel tetrazole-containing phosphonoacetamidines – 2-[(N-tert-butyl- or N-adamantyl)imino-2-(tetrazolyl)ethyl]phosphonic acid dialkyl esters – with good yields. The reactions occur via nucleophilic attack of the tetrazole N-1 atom at the C=C bond. | |
| 49 | K.S. Rodygin, V.P. Ananikov, “An efficient metal-free pathway to vinyl thioesters with calcium carbide as the acetylene source”, Green Chemistry 2015, accepted DOI:10.1039/c5gc01552a | |
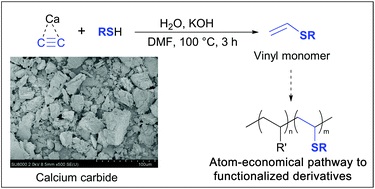 |
Abstract. Chemical reactions involving high-pressure acetylene are not easily performed in a standard laboratory setup. The risk of explosion and technical difficulties drastically complicate the equipment and greatly increase the cost. In this study, we propose the replacement of acetylene with calcium carbide, which was successfully utilized to synthesize practically useful vinyl thioesters in accordance with a simple and environmentally benign procedure. The reaction proceeded under mild conditions using a standard laboratory setup. The optimized reaction conditions allowed the selective synthesis of the vinyl thioesters in high yields, and the reaction conditions can be scaled up to synthesize grams of sulfides from inexpensive starting materials. | |
| 50 | E.E. Galenko, O.A. Tomashenko, A.F. Khlebnikov, M.S. Novikov, “Fe(II)/Et3N-Relay-catalyzed domino reaction of isoxazoles with imidazolium salts in the synthesis of methyl 4-imidazolylpyrrole-2-carboxylates, its ylide and betaine derivatives”, Beilstein J. Org. Chem 2015, 11, 1732-1740 DOI:10.3762/bjoc.11.189 | |
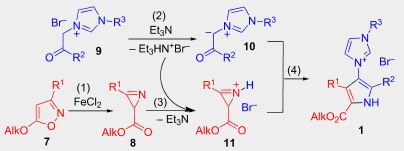 |
Abstract. A simple approach was developed for the synthesis of methyl 4-imidazolylpyrrole-2-carboxylates from easily available compounds, 5-methoxyisoxazoles and phenacylimidazolium salts under hybrid Fe(II)/Et3N relay catalysis. The products were easily transformed into the corresponding 3-(5-methoxycarbonyl-1H-imidazol-3-ium-3-yl)pyrrol-1-ides, which in turn can be hydrolyzed under basic conditions into the corresponding betaines. A carbene tautomeric form of the 4-methoxycarbonyl-substituted imidazolylpyrrolides was trapped by reaction with sulfur affording the corresponding imidazolethiones under very mild conditions. | |
| 51 | O.A. Tomashenko, A.F. Khlebnikov, I.P Mosiagin, M.S. Novikov, E.V. Grachova, J.R. Shakirova, S.P. Tunik, “A new heterocyclic skeleton with highly tunable absorbtion/emission wavelength via H-bonding”, RSC Adv. 2015, accepted DOI:10.1039/C5RA17755C | |
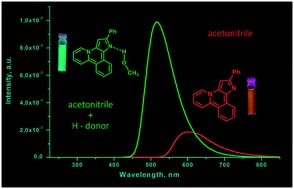 |
Abstract. A new heterocyclic system, pyrido[2,1-a]pyrrolo[3,2-c]isoquinoline, was synthesized via Pd-catalyzed intramolecular cyclization of 1-[1-benzyl-2-(2-bromophenyl)-1H-pyrrol-3-yl]pyridin-1-ium bromides. The heterocycles obtained display stimuli responsive fluorescence in solution depending on the nature of the solvent. The strongest blue shift of the emission maxima and growth in luminescence intensity was observed in protic solvents and upon addition of proton donors to solutions of compounds in aprotic solvents. The effect of proton donors on emission characteristics was explained by DFT calculations in terms of H-complex formation with the nucleophilic centres of the molecular skeleton. | |
| 52 | N.A. Danilkina, A.G. Lyapunova, A.F. Khlebnikov, G.L. Starova, S. Bräse, I.A. Balova, “Ring-Closing Metathesis of Co2(CO)6–Alkyne Complexes for the Synthesis of 11-Membered Dienediynes: Overcoming Thermodynamic Barriers”, J. Org. Chem. 2015, 80, 5546-5555 DOI:10.1021/acs.joc.5b00409 | |
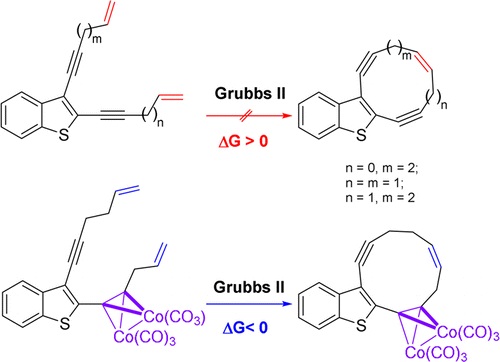 |
Abstract. The feasibility of ring-closing metathesis (RCM) as a synthetic entry to 10- and 11-membered dienediynes fused to a benzothiophene core was explored by experimental and theoretical investigations. An established sequence of iodocyclization of o-(buta-1,3-diynyl)thioanisoles followed by Sonogashira coupling to form diethynylbenzothiophenes was used to synthesize terminal benzothiophene-fused enediyne diolefins as substrates for RCM. Encountering an unexpected lack of reactivity of these substrates under standard RCM conditions, we applied DFT calculations to reveal that the underlying cause was a positive change in Gibbs free energy. The change in Gibbs free energy was also found to be positive for RCM of indole- and benzannulated terminal diolefins when affording smaller than 12-membered rings. We found that modification of the enediyne–diolefin substrate as the Co2(CO)6–alkyne complex allowed the target benzothiophene-fused 11-membered dienediyne to be obtained via RCM; the alkyne complexation strategy therefore provides one valid technique for overcoming challenges to macrocyclization of this kind. | |
| 53 | A.V. Galenko, A.F. Khlebnikov, M.S. Novikov, M.S. Avdontseva, “Synthesis of 3-(1,2-dioxoethyl)- and 2,3-dicarbonyl-containing pyrroles”, Tetrahedron 2015, 71, 1940-1951 DOI:10.1016/j.tet.2015.02.030 | |
 |
Abstract. The transition metal-catalyzed reaction of 2H-azirines with 1,2,4-tricarbonyl compounds leads to 3-(1,2-dioxoethyl)- and 2,3-dicarbonyl-pyrrole derivatives, useful building blocks for the preparation of 3-heterocyclyl pyrroles and pyrroles fused with heterocycles. The influence of catalysts and the reaction conditions on the yields of pyrroles and the regioselectivity of the reaction were studied. Experimental and theoretical results suggest that the reaction proceeds via the formation of an intermediate azirine–metal complex and subsequent nucleophilic N–C3 bond cleavage. | |
| 54 | N.V. Rostovskii, P.A. Sakharo, M.S. Novikov, A.F. Khlebnikov, G.L. Starova, “Cu(I)-NHC-Catalyzed (2+3)-Annulation of Tetramic Acids with 2H-Azirines: Stereoselective Synthesis of Functionalized Hexahydropyrrolo[3,4-b]pyrroles”, Org. Lett. 2015, 17, 4148-4151 DOI:10.1021/acs.orglett.5b01883 | |
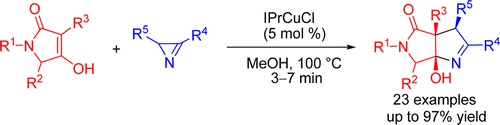 |
Abstract. A stereoselective and high-yield synthesis of hexahydropyrrolo[3,4-b]pyrroles from tetramic acids and 2H-azirines under Cu(I)–NHC catalysis is developed. An unusual N–C2 azirine bond cleavage, initiated by a copper enolate, was rationalized in terms of a free radical reaction mechanism. | |
| 55 | E.E. Galenko, O.A. Tomashenko, A.F. Khlebnikov, M.S. Novikov, “Metal/organo relay catalysis in a one‐pot synthesis of methyl 4aminopyrrole‐2‐carboxylates from 5‐methoxyisoxazoles and pyridinium ylides”, Org. Biomolec. Chem. 2015, 13, 9825-9833 DOI:10.1039/c5ob01537e | |
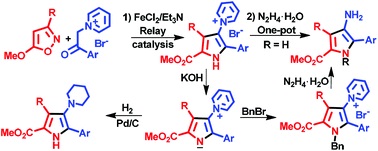 |
Abstract. Methyl 4-aminopyrrole-2-carboxylates were synthesized in one-pot mode by the relay catalytic cascade reaction of 5-methoxyisoxazoles with pyridinium ylides by the use of a FeCl2/Et3N binary catalytic system leading to 1-(5-methoxycarbonyl-1H-pyrrol-3-yl)pyridinium salts followed by hydrazinolysis. The approach permits the introduction of a substituent at the pyrrole nitrogen via a nucleophilic reaction of the pyrrolylpyridinium ylide derived from the salt. Catalytic reduction of the ylides gives methyl 4-piperidinopyrrole-2-carboxylates. | |
| 56 | E.E. Galenko, A.V. Galenko, A.F. Khlebnikov, M.S. Novikov, “Domino transformation of isoxazoles to 2,4-dicarbonylpyrroles under Fe/Ni relay catalysis”, RSC Adv. 2015, 5, 18172-18176 DOI:10.1039/C5RA01889G | |
 |
Abstract. The domino reaction of 5-alkoxy- or 5-aminoisoxazoles, under metal relay catalysis, with symmetric 1,3-diketones gives 4-acylpyrrole-2-carboxylic acid derivatives in high yield. Esters and amides of acylacetic acids react regioselectively, giving derivatives of pyrrole-2,4-dicarboxylic acid as the main products. | |
| 57 | Е.А. Катленок, А.А. Золотарев, А.Ю. Иванов, С.Н. Смирнов, Р.И. Байчурин, К.П. Балашев, «Строение, оптические и электрохимические свойства циклометаллированных комплексов иридия с 2-фенилбензотиазолом и n-(бензотиазол)-n-идоацетамидинат- и n-(тиазол)-n-идоацетамидинат-ионами», Журнал Общей Химии 2015, 85, 1896-1902 | |
| 58 | А.В. Бутлак, Ю.В. Кондратьев, А.С. Мазур, А.Ю. Тимошкин, «Термическое разложение амминборана при 357 K», Журнал Общей Химии 2015, 85, 1761-1764 | |
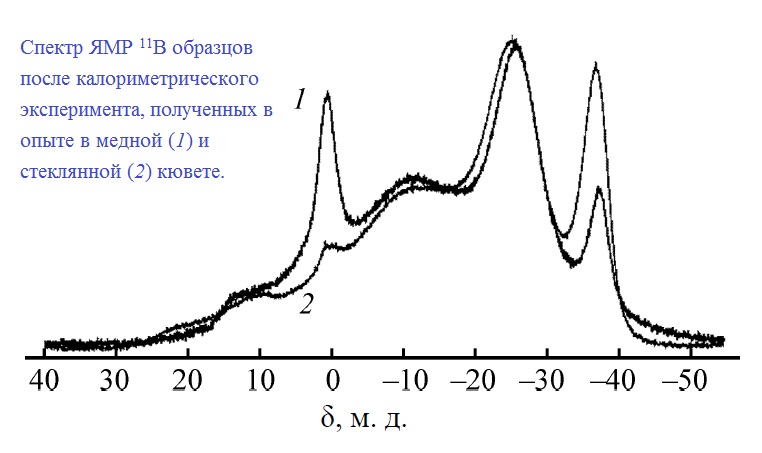 |
Abstract. Калориметрическим методом определены тепловые эффекты разложения амминборана при 357 K и атмосферном давлении в стеклянных и медных кюветах. Энтальпия термического разложения BH3NH3 в стеклянных ампулах составляет –24.8±2.3 кДж/моль, эта величина вклю- чает структурную перестройку в диаммониат диборана –4.0±2.8 кДж/моль и тепловой эффект разложения до полиамидоборана –20.8±1.6 кДж/моль. В случае опытов в медных кюветах наблюдается существенно больший (–47.6±8.3 кДж/моль) экзотермический эффект. Анализ образцов после калориметрических опытов методом твердотельного ЯМР на ядрах 15N и 11B показал, что в ходе опытов в медных кюветах происходит частичное окисление с образованием борной кислоты. |
|
| 59 | Е.А. Катленок, А.А. Золотарев, А.Ю. Иванов, С.Н. Смирнов, К.П. Балашев, «Строение, оптические и электрохимические свойства биядерных комплексов с платинированным 2-фенилбензотиазолом, и мостиковыми 2-меркапто-производными пиридина, пиримидина, бензотиазола и бензоксазола», Журнал Структурной Химии 2015, 56(5), 937-944 | |
| 60 | A.D. Lisakova, D.S. Ryabukhin, R.E. Trifonov, V.A. Ostrovskii, A.V. Vasilyev, “Alkylation of 5-substituted NH-tetrazoles by alcohols in the superacid CF3SO3H”, Tetrahedron Lett. 2015, 56, 7020-7023 DOI:10.1016/j.tetlet.2015.11.005 | |
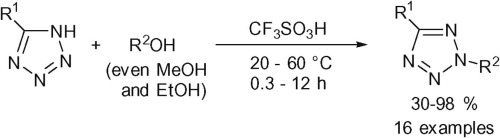 |
Abstract. Reactions of 5-substituted NH-tetrazoles with alcohols in the superacid CF3SO3H have been studied. Both the structure of the tetrazole and the nature of alcohol were found to dramatically influence the selectivity of the reaction and yields of products. Tetrazoles bearing phenyl, electron-donating aryl, or benzyl groups at the 5-position, have been alkylated using various alcohols (including MeOH and EtOH) in CF3SO3H upon heating at 60 °C for 0.3–12 h to afford 2-alkyl-2H-tetrazoles in 30–98% yields. | |
| 61 | А.Д. Лисакова, Д.С. Рябухин, Р.Е. Трифонов, В.А. Островский, А.В. Васильев, «Гидроарилирование (E)-2-метил-5-(2-фенилэтен-1-ил)-2Н-тетразола в условиях суперэлектрофильной активации», Журнал органической химии,2015, 9, 1382-1384 | |
| 62 | Р.О. Яковенко, В.М. Музалевский, В.Г. Ненайденко, А.В. Васильев, «Реакции 4-фенил-1,1,1-трифторбут-3-ен-2-она с аренами в суперкислоте CF3SO3H», Журнал органической химии, 2015, 3, 449-451 | |
| 63 | D.N. Zakusilo, D.S. Ryabukhin, I.A. Boyarskaya, O.S. Yuzikhin, A.V. Vasilyev, «Tandem superelectrophilic hydroarylation of C=C bond and carbonyl reduction in cinnamides: synthetic rout to 3,3-diarylpropylamines, valuable pharmaceuticals», Tetrahedron, 2015, 71, 102-108 DOI:10.1016/j.tet.2014.11.033 | |
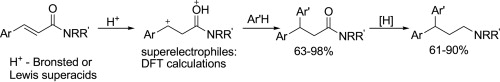 |
Abstract. Cinnamides ArCHdouble bond; length as m-dashCHCONRR′ in reactions with arenes Ar′H under the action of Bronsted (TfOH, FSO3H) or Lewis (AlBr3) superacids at rt for 1–2 h give Cdouble bond; length as m-dashC bond hydroarylation products ArAr′CHCH2CONRR′ in yields of 63–98%. Reduction (LiAlH4/Et2O) of carbonyl group in the latter results in the formation of 3,3-diarylpropylamines ArAr′CHCH2CH2NRR′, valuable drugs. The reaction intermediates, superelectrophilic dications ArC+H–CH2C(OH+)NRR′, have been characterized by DFT calculations in terms of global electrophilicity index, natural charges, and atomic orbitals contributions. | |
| 64 | D.S. Ryabukhin, A.V. Vasilyev, «A synthesis of 4-aryl quinolin-2(1H)-ones by acidic zeolite promoted intramolecular cyclization of N-aryl amides of 3-arylpropynoic acids», Tetrahedron Lett. 2015, 56, 2200-2202 DOI:10.1016/j.tetlet.2015.03.060 | |
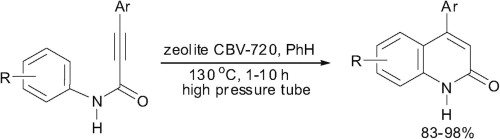 |
Abstract. N-Aryl amides of 3-arylpropynoic acids are intramolecularly cyclized into 4-aryl quinolin-2(1H)-ones in high yields without any by-products under the action of acidic zeolite CBV-720 in benzene at 130 °C in a glass high pressure tube. | |
| 65 | A.A. Beljaev, D.V. Krupenya, E.V. Grachova, V.V. Gurzhiy, A.S. Melnikov, P.Yu. Serdobintsev, E.S. Sinitsyna, E.G. Vlakh, T.B. Tennikova, S.P. Tunik, «Supramolecular AuI-CuI complexes as new luminescent labels for covalent bioconjugation», Bioconjugate Chem. 2015, ASAP DOI:10.1021/acs.bioconjchem.5b00563 | |
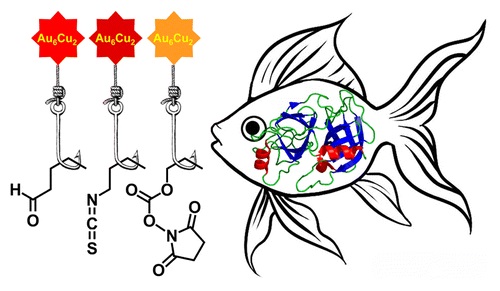 |
Abstract. Two new supramolecular organometallic complexes, namely, [Au6Cu2(C2C6H4CHO)6(PPh2C6H4PPh2)3](PF6)2 and [Au6Cu2(C2C6H4NCS)6(PPh2C6H4PPh2)3](PF6)2, with highly reactive aldehyde and isothiocyanate groups have been synthesized and characterized using X-ray crystallography, ESI mass spectrometry, and NMR spectroscopy. The compounds obtained demonstrated bright emission in solution with the excited-state lifetime in microsecond domain both under single- and two-photon excitation. The luminescent complexes were found to be suitable for bioconjugation in aqueous media. In particular, they are able to form the covalent conjugates with proteins of different molecular size (soybean trypsin inhibitor, human serum albumin, rabbit anti-HSA antibodies). The conjugates demonstrated a high level of the phosphorescent emission from the covalently bound label, excellent solubility, and high stability in physiological media. The highest quantum yield, storage stability, and luminance were detected for bioconjugates formed by covalent attachment of the aldehyde-bearing supramolecular AuI–CuI complex. The measured biological activity of one of the labeled model proteins clearly showed that introduced label did not prevent the biorecognition and specific protein–protein complex formation that was extremely important for the application of the conjugates in biomolecular detection and imaging. | |
| 66 | Yu. Kondratev, A. Butlak, I. Kazakov, A. Timoshkin, «Sublimation and thermal decomposition of ammonia borane: Competitive processes controlled by pressure», Thermochim. Acta 2015, ASAP DOI:10.1016/j.tca.2015.08.021 | |
 |
Abstract. Thermal behavior of ammonia borane BH3NH3 (AB) has been studied by calorimetry, tensimetry and mass spectrometry methods. It is shown, that depending on vapor pressure in the system two competitive processes are taking place at 357 K. At atmospheric pressure thermal decomposition with hydrogen evolution is the dominant process: BH3NH3(s) = 1/n (BH2NH2)n(s) + H2(g) (1). At low pressures (circa 4 mTorr) the major process is endothermic sublimation of AB: BH3NH3(s) = BH3NH3(g) (2). At intermediate pressures both processes occur simultaneously. Enthalpies for the processes (1) and (2) have been determined by drop-calorimetry method: Δ(1)H357° = −24.8 ± 2.3 kJ mol−1 and ΔsubH357°(BH3NH3) = 76.3 ± 3.0 kJ mol−1. Solid products after sublimation and decomposition have been characterized by IR and NMR spectroscopy; gaseous forms were studied by mass spectrometry. Activation energy of 94 ± 11 kJ mol−1 for the process (1) in range 327–351 K was determined by static tensimetry method. Based on the analysis of available thermodynamic characteristics, new values for the standard formation enthalpy of solid AB −133.4 ± 5.2 kJ mol−1 and polyamidoborane −156.7 ± 5.8 kJ mol−1 are recommended. | |
| 67 | A.S. Smirnov, E.S. Yandanova, N.A. Bokach, G.L. Starova, V.V. Gurzhiy, M.S. Avdontceva, A.A. Zolotarev, V.Yu. Kukushkin, «Zinc(II)-mediated generation of 5-amino substituted 2,3-dihydro-1,2,4-oxadiazoles and their further ZnII-catalyzed and O2-involving transformations», New J. Chem. 2015, 12, 9330-9344 DOI:10.1039/C5NJ02061A | |
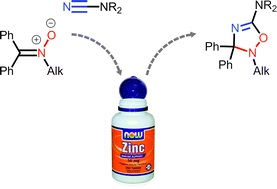 |
Abstract. ZnII-activated cyanamides NCNR2 (R2 = Me2, Et2, C5H10, (CH2)2O(CH2)2, Ph2) react with the acyclic N-alkyl ketonitrones Ph2C[double bond, length as m-dash]N+(O−)R′ (R′ = Me, CH2Ph) and N-aryl ketonitrones (R′ = Ph, p-BrC6H4, p-EtC6H4) under mild conditions. Uncomplexed 5-aminosubstituted 2,3-dihydro-1,2,4-oxadiazoles (6 examples; 49–82%) were obtained in zinc(II)-involving cycloaddition of the N-alkyl ketonitrones to the cyanamide substrates; these 2,3-dihydro-1,2,4-oxadiazoles undergo ring-opening giving carbamoylamidines and methylidenureas. The N-aryl ketonitrones react with ZnII-activated cyanamides giving the open-chain systems, viz. carbamoylamidines, N′-(2-(diphenylmethylidene)amino)-phenyl-N,N-carbamimidic acids, and methylidenureas, which are presumably formed via the cycloaddition route followed by the N–O cleavage induced by the acceptor character of the aryl groups. | |
| 68 | I.S. Krytchankou, I.O. Koshevoy, V.V. Gurzhiy, V.A. Pomogaev, S.P. Tunik, «Luminescence Solvato- and Vapochromism of Alkynyl-Phosphine Copper Clusters», Inorg. Chem. 2015, 17, 8288–8297 DOI:10.1021/acs.inorgchem.5b00239 | |
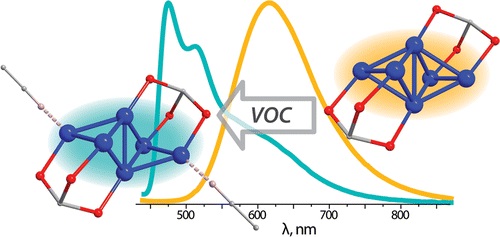 |
Abstract. The reaction of [Cu(NCMe)4][PF6] with aromatic acetylenes HC2R and triphosphine 1,1,1-tris(diphenylphosphino)methane in the presence of NEt3 results in the formation of hexanuclear Cu(I) clusters with the general formula [Cu6(C2R)4{(PPh2)3CH}2][PF6]2 (R = 4-X-C6H4 (1–5) and C5H4N (6); X = NMe2 (1), OMe (2), H (3), Ph (4), CF3 (5)). The structural motif of the complexes studied consists of a Cu6 metal core supported by two phosphine ligands and stabilized by σ- and π-coordination of the alkynyl fragments (together with coordination of pyridine nitrogen atoms in cluster 6). The solid state structures of complexes 2–6 were determined by single crystal XRD analysis. The structures of the complexes in solution were elucidated by 1H, 31P, 1H–1H COSY NMR spectroscopy, and ESI mass spectrometry. Clusters 1–6 exhibit moderately strong phosphorescence in the solid state with quantum yields up to 17%. Complexes 1–5 were found to form solvates (acetone, acetonitrile) in the solid state. The coordination of loosely bound solvent molecules strongly affects emission characteristics and leads to solvato- and vapochromic behavior of the clusters. Thus, solvent-free and acetonitrile solvated forms of 3 demonstrate contrasting emission in orange (615 nm) and blue (475 nm) regions, respectively. The computational studies show that alkynyl-centered IL transitions mixed with those of MLCT between the Cu6 metal core and the ligand environment play a dominant role in the formation of excited states and can be considerably modulated by weakly coordinating solvent molecules leading to luminescence vapochromism. | |
| 69 | M.Ya. Demakova, D.S. Bolotin, N.A. Bokach, R.M. Islamova, G.L. Starova, V.Yu. Kukushkin, «Click-Type PtII-Mediated Hydroxyguanidine–Nitrile Coupling Provides Useful Catalysts for Hydrosilylation Cross-Linking», ChemPlusChem 2015, 80, 1607-1614 DOI:10.1002/cplu.201500327 | |
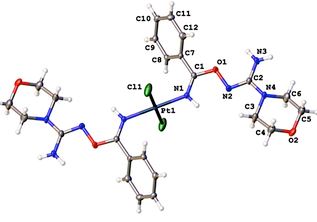 |
Abstract. The nitrile complexes trans-[PtCl2(RCN)2] (R=Et (NC1), tBu (NC2), Ph (NC3), p-BrC6H4 (NC4)) and cis-[PtCl2(RCN)2] (R=Et (NC5), tBu (NC6), Ph (NC7)) react with 1 equiv of the hydroxyguanidine OC4H8NC(=NOH)NH2 (HG) furnishing the mono-addition products trans- and cis-[PtCl2(RCN){NH=C(R)ON=C(NH2)NC4H8O}] (1–4 and 9–11; 7 examples; 54–74 % yield). Treatment of any of the nitrile complexes NC1–NC7 with HG in a 1:2 molar ratio generated the bis-addition products trans- and cis-[PtCl2{NH=C(R)ON=C(NH2)NC4H8O}2] (5–8 and 12–14; 7 examples; 69–89 % yield). The PtII-mediated coupling between nitrile ligands and HG proceeds substantially faster than the corresponding reactions involving amid- and ketoximes and gives redox stable products under normal conditions. Complexes 1, 6⋅4 CH2Cl2, 7⋅4 CH2Cl2, 8⋅2 CH2Cl2, and NC4 were studied by X-ray crystallography. Platinum(II) species 1–3, 10, 11, and especially 9, efficiently catalyze the hydrosilylation cross-linking of vinyl-terminated poly(dimethylsiloxane) and trimethylsilyl-terminated poly(dimethylsiloxane-co-ethylhydrosiloxane) giving high-quality thermally stable silicon resins with no structural defects. The usage of these platinum species as the catalysts does not require any inhibitors and, moreover, the complexes and their mixtures with vinyl- and trimethylsilyl-terminated polysiloxanes are shelf-stable in air. | |
| 70 | K.S. Rodygin, A.A. Kostin, V.P. Ananikov, «Calcium carbide as a convenient acetylene sourse in the synthesis of unsarurated sulfides, promising functionalized monomers», Mendeleev Commun. 2015, 25, 415-416 DOI:10.1016/j.mencom.2015.11.004 | |
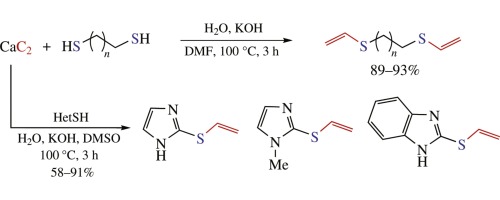 |
Abstract. Calcium carbide was studied as a useful solid-state reagent to incorporate acetylene unit into synthetic procedures. Atom-economic thiol-yne click reaction was successfully performed with single and double additions. Heterocyclic thiols and aliphatic dithiols reacted with acetylene generated in situ from calcium carbide to afford corresponding vinyl sulfides and bis(thiovinyl)ethers in good to high yields. | |
| 71 | E. Abakumov, E. Lodygin, V. Tomashunas, «13C NMR and ESR acterization of Humic Substances Isolated Soils of Two Siberian Arctic Islands», Int. J. Ecology 2015, 7, 1-10 DOI:DOI:10.1155/2015/390591 | |
 |
Abstract. Humic acids (HAs) and fulvic acids (FAs) of two Polar soils were investigated by 13C NMR and ESR spectroscopies, investigating the degree of humification and the molecular structure. One soil, from Bolshoi Lyakhovsky Island, contains two humus horizons: modern and buried. The other soil, from Wrangel Island, had only one modern humus horizon. The HAs and FAs of the two soils investigated show essential differences. The HAs show fewer oxygen-containing groups in comparison with the FAs, whereas the degree of aromaticity is two or three times higher in the HAs. The 13C NMR data also show that HAs are very different from FAs in terms of their molecular composition and hydrophobicity. Humification in the Arctic is limited by the very low content of lignin-derived compounds, due to the restricted vascular flora. As a result, the HAs, isolated from Polar soils, are more similar to the corresponding FAs than to the typical HAs of temperate soils. This was confirmed by ESR data, which show similar levels of free radical concentration for HAs and FAs and are related to the low level of aromaticity of both materials investigated. Apparently, the humification process in the soils of Polar Arctic deserts is in an initial stage. | |
| 72 | E. Ejarque, E. Abakumov «Stability and biodegradability of humic substances Arctic soils of Western Siberia: insights 13C-NMR spectroscopy and elemental analysis», Solid Earth Discussion 2015, 7, 3021-3052 DOI:10.5194/sed-7-3021-2015 | |
 |
Abstract. Arctic soils contain large amounts of organic matter which, globally, exceed the amount of carbon stored in vegetation biomass and in the atmosphere. Recent studies emphasise the potential sensitivity for this soil organic matter (SOM) to be mineralised when faced with increasing ambient temperatures. In order to better refine the predictions about the response of SOM to climate warming, there is a need to increase the spatial coverage of empirical data on SOM quantity and quality in the Arctic area. This study provides, for the first time, a characterisation of SOM from the Gydan Peninsula in the Yamal Region, Western Siberia, Russia. On the one hand, soil humic acids and their humification state were characterised by measuring the elemental composition and diversity of functional groups using solid-state 13C-nuclear magnetic resonance (NMR) spectroscopy. Also, the total mineralisable carbon was measured. Our results indicate that there is a predominance of aliphatic carbon structures, with a minimal variation of their functional-group composition both regionally and within soil depth. This vertical homogeneity and low level of aromaticity reflects the accumulation in soil of lowly decomposed organic matter due to cold temperatures. Mineralisation rates were found to be independent of SOM quality, and to be mainly explained solely by the total carbon content. Overall, our results provide further evidence on the sensitivity that the soils of Western Siberia may have to increasing ambient temperatures and highlight the important role that this region can play in the global carbon balance under the effects of climate warming. | |
| 73 | I.A. Smetanin, M.S. Novikov, N.V. Rostovskii, A.F. Khlebnikov, G.L. Starova, D.S. Yufit, «4-Halo-2-azabuta-1,3-dienes as intermediates in the rhodium carbenoid-initiated transformation of 2-halo-2H-azirines into 2,3-dihydroazetes and 2,5-dihydrooxazoles», Tetrahedron 2015, 71, 4616-4628 DOI:org/10.1016/j.tet.2015.05.022 | |
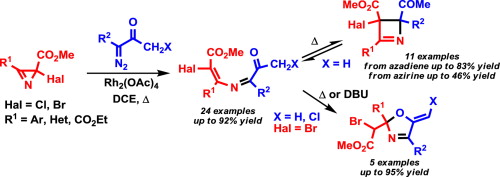 |
Abstract. A wide range of electron-poor 4-bromo-/4-chloro-2-azabuta-1,3-dienes were synthesized by the Rh2(OAc)4-catalyzed reaction of diazo esters and diazo ketones with methyl 2-halo-2H-azirine-2-carboxylates. The E stereoselectivity with respect to the configuration of the Cdouble bond; length as m-dashC bond of the 2-azadiene moiety is in good agreement with the results of DFT (M06-2X/6-31+G(d,p)) calculations of the reaction pathway. The reaction proceeds via the formation of an azirinium ylide intermediate followed by ring opening with outward rotation of the halogen atom. Depending on the substitution pattern at C1 electron-poor 4-halo-2-azabutadienes can undergo two types of cyclization at elevated temperatures: reversible 1,4-electrocyclization to give 2,3-dihydroazetes or 1,5-exo-trig cyclization to give 5-methylene-2,5-dihydrooxazoles, both in good yields. The dihydrooxazole derivatives can be also obtained at ambient temperature under DBU catalysis. | |
| 74 | S.A. Kras’ko, S.S. Zlotskii, V.P. Boyarskii, “Comparative Activity of Aryl, Alkyl, and Cycloalkyl Halides in the Suzuki Reaction Catalyzed with Acyclic Diaminocarbene Complex of Palladium”, Russ. J. Org. Chem. 2015, 85, 2541-2546 DOI:10.1134/S1070363215110079 | |
 |
Abstract. Relative activity of halogenated arenes, alkanes, and alkanes in the Suzuki reaction catalyzed with acyclic diaminocarbene complex of palladium has been investigated. Under all the investigated conditions, 4-iodoanisole has been more active than the alkyl halides. The reaction with 4-methyl-1-(chloromethyl)benzene has afforded the target 4-methyl-1-(phenylmethyl)benzene along with significant amount of by-products; other alkyl and cycloalkyl halides do not participate into the cross-coupling reaction. Ethanol has been found the most suitable solvent for the reaction. The reaction in acetonitrile provides noticeable yield of the products only in the presence of polyethylene glycol and water. | |
| 75 | D.V. Kurandina, E.V. Eliseenkov, T.Sh. Khaibulova, A.A. Petrov, V.P. Boyarskii, “Effect of the Structural Factors on Reactivity of Aryl Halides in the Copper-Catalyzed Arylation of Aniline in Aqueous Medium”, Russ. J. Gen. Chem. 2015, 85, 2277-2281 DOI:10.1134/S1070363215100096 | |
 |
Abstract. Arylation of aniline with substituted bromobenzenes in two-phase water–chlorobenzene system catalyzed by Cu(OAc)2–N-phenyloxalyldihydrazide–hexane-2,5-dione system was found to be slightly sensitive to the polar effect of a substituent in the aryl halide. Value of the reaction parameter ρ was 0.55. However the presence of substituent in ortho-position of the benzene ring decreased significantly the reaction rate [k rel(2-MeC6H4Br/4-MeC6H4Br) = 0.14.] | |
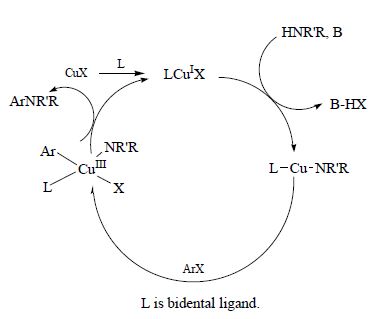
| 76 | V.A. Rassadin, V.P. Boyarskiy, V.Yu. Kukushkin, “Facile Gold-Catalyzed Heterocyclization of Terminal Alkynes and Cyanamides Leading to Substituted 2-Amino-1,3-Oxazoles”, Org. Lett. 2015, 17, 3502-3505 DOI:10.1021/acs.orglett.5b01592 | |
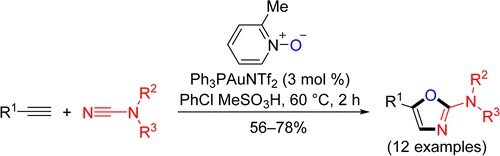 |
Abstract. Facile gold-catalyzed heterocyclization based upon intermolecular trapping of the generated α-oxo gold carbenes with various cyanamides R2R3NCN (R2/R3 = Alk/Alk, −(CH2)2O(CH2)2–, Ar/Ar, Ar/H) has been developed. In most cases, 2-amino-1,3-oxazoles functionalized at the nitrogen atom as well as at the fifth position of the heterocyclic ring (12 examples) were isolated in good to moderate yields. | |
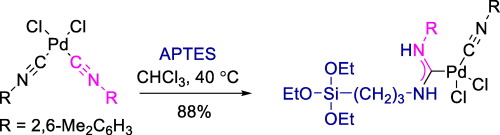
| 77 | V.A. Rassadin, A.A. Yakimanskiy, E.V. Eliseenkov, V.P. Boyarskiy, “Synthesis of acyclic diaminocarbene palladium complex featuring triethoxysilane moiety”, Inorg. Chem. Comm. 2015, 61, 21-23 DOI:10.1016/j.inoche.2015.08.008 | |
 |
Abstract. The first example of an acyclic diaminocarbene palladium complex, viz. cis-[PdCl2(CN(2,6-Me2C6H3)){C(NH(2,6-Me2C6H3)) = NH(CH2)3Si(OEt)3}], featuring the triethoxysilane moiety is described. The complex was generated from bis(o-xylylisocyanide)palladium dichloride and (3-aminopropyl)triethoxysilane under mild conditions and isolated in 88% yield. The target compound is stable at RT either in the solid state or in CDCl3 or CD3OD solutions within several months. | |
| 78 | D.S. Bolotin, M.Ya. Demakova, A.S. Novikov, M.S. Avdontceva, M.L. Kuznetsov, N.A. Bokach, V.Yu. Kukushkin, “Bifunctional Reactivity of Amidoximes Observed upon Nucleophilic Addition to Metal-Activated Nitriles”, Inorg. Chem. 2015, 54, 4039-4046 DOI:10.1021/acs.inorgchem.5b00253 | |
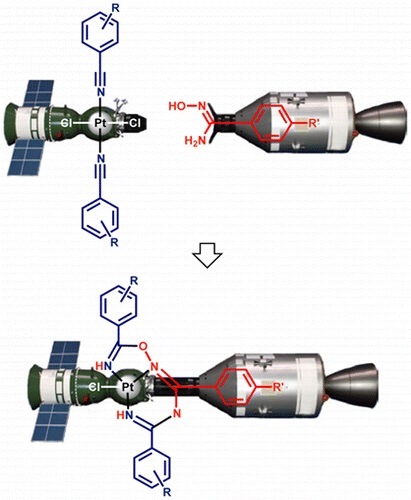 |
Abstract. Treatment of the aromatic nitrile complexes trans-[PtCl2(RC6H4CN)2] (R = p-CF3 NC1, H NC2, o-Cl NC3) with the aryl amidoximes p-R′C6H4C(NH2)═NOH (R′ = Me AO1, H AO2, Br AO3, CF3 AO4, NO2 AO5) in all combinations, followed by addition of 1 equiv of AgOTf and then 5 equiv of Et3N, leads to the chelates [PtCl{HN═C(RC6H4)ON═C(C6H4R′-p)NC(RC6H4)═NH}] (1–15; 15 examples; yields 71–88% after column chromatography) derived from the platinum(II)-mediated coupling between metal-activated nitriles and amidoximes. The mechanism of this reaction was studied experimentally by trapping and identification of the reaction intermediates, and it was also investigated theoretically at the DFT level of theory. The combined experimental and theoretical results indicate that the coupling with the nitrile ligands involves both the HON and monodeprotonated NH2 groups of the amidoximes, whereas in the absence of the base, the NH2 functionality is inactive toward the coupling. The observed reaction represents the first example of bifunctional nucleophilic behavior of amidoximes. The complexes 1–16 were characterized by elemental analyses (C, H, N), high-resolution ESI+-MS, FTIR, and 1H NMR techniques, whereas unstable 17 was characterized by HRESI+-MS and FTIR. In addition, 8·C4H8O2, 12, and 16·CHCl3 were studied by single-crystal X-ray diffraction. | |
| 79 | N.A. Danilkina, P.S. Vlasov, S.M. Vodianik, A.A. Kruchinin, Y.G. Vlasov, I.A. Balova, “Synthesis and chemosensing properties of cinnoline-containing poly(arylene ethynylene)s”, Beilstein J. Org. Chem. 2015, 11, 373-384 DOI:10.3762/bjoc.11.43 | |
 |
Abstract. Novel poly(arylene ethynylene)s comprising a cinnoline core were prepared in high yields via a three-step methodology. A Richter-type cyclization of 2-ethynyl- and 2-(buta-1,3-diynyl)aryltriazenes was used for cinnoline ring formation, followed by a Sonogashira coupling for the introduction of trimethylsilylethynyl moieties and a sila-Sonogashira coupling as the polycondensation technique. The fluorescence of the cinnoline-containing polymers in THF was highly sensitive to quenching by Pd2+ ions. | |
| 80 | S.S. Chuprun, E.A. Popova, A.V. Mukhametshina, R.E. Trifonov, “Synthesis of Tetrazol-1-yl Analogs of L-Lysine and L-Ornithine”, Russ. J. Org. Chem. 2015, 51, 1671-1673 DOI:10.1134/S1070428015110287 | |
 |
||
| 81 | A.S. Kaledina, A.D. Zorina, V.V. Anokhina, R.E. Trifonov, “Synthesis of Tetrazol-5-ylethyl Derivatives of Dipterocarpol”, Russ. J. Org. Chem. 2015, 51, 1674-1675 DOI:10.1134/S1070428015110299 | |
 |
||
| 82 | D. Dar’In, O. Bakulina, M. Chizhova, M. Krasavin, “New Heterocyclic Product Space for the Castagnoli-Cushman Three-Component Reaction”, Organic Lett. 2015, 17, 3930-3933 DOI:10.1021/acs.orglett.5b02014 | |
 |
Abstract. Significant expansion of heterocyclic product space accessible by the Castagnoli–Cushman reaction (CCR) has been achieved via the use of glutaric anhydride analogues containing endocyclic substitutions with oxygen, nitrogen, and sulfur. Incorporation of these heteroatoms in the anhydride’s backbone results in enhanced reactivity and generally lower temperatures that are required for the reactions to go to completion. These findings are particularly significant in light of the CCR recently recognized as an efficient tool for lead-oriented synthesis. | |
The table above lists Articles acknowledging the Center.